Aldol-like Reactions
Introduction
The aldol reaction is one of the most useful means for forming C–C bonds. It can join two substrates and create two stereocenters. The product has multiple functional groups that permit further chemistry to be performed. The concepts underlying the aldol reaction are even more useful to an undergraduate chemist. The idea that an enolizable functional group (or equivalent) can be transformed into a nucleophile that will attack an electrophilic carbonyl group, or equivalent functionality, is the basis of many named reactions. A generalization of this concept is given below:

A generalization of aldol-like reactions that involve the addition of a nucleophile, formed by the enolization of a suitable functional group, to a carbonyl-containing compound, or its equivalent (in which case it technically isn’t an enolization but the principle is the same). The reaction will result in the formation of a C–C bond and possibly a C=C bond. It will leave the original functional group of the nucleophile unchanged but will lead to the electrophile accepting electrons.
In this summary, I will cover a number of the more common examples. Each one of these reactions has the same fundamental mechanism, it is just the decoration that differs (the atoms surrounding the reacting functionality). There are many more examples that I will not cover. If you like these reactions, simply pick up a textbook entitled “Named Organic Reactions” and there will be 10s of examples. Personally, I don’t think the names are important, it is the mechanism that is key as this can be applied to all the examples!
The Claisen Condensation
The aldol addition involved the reaction of aldehydes and/or ketones. Nucleophilic addition occurred to a carbonyl group and resulted in the formation of an alcohol (or alkoxide). For more information about addition to aldehydes and ketones look HERE and HERE. If the electrophile is changed to an ester then there is a leaving group attached to the carbonyl group. This alters the course of the reaction and the product will now be a ketone.

The Claisen Condensation of two identical esters. The reaction leads to the formation of a β-keto ester. Standard keto-enol tautomerization favors the compound existing as the conjugated enol form (see HERE for a discussion of this phenomenon).
The mechanism of the reaction should be predictable. It follows the same path as the aldol reaction except that the leaving group on the carbonyl group permits the tetrahedral intermediate to collapse to give a ketone. This is shown below:

The mechanism of the Claisen condensation involves enolization of carbonyl-containing functional group (normally aldehyde, ketone or ester) followed by nucleophilic addition to an ester. The resulting tetrahedral intermediate collapses, kicking out an alkoxide. Up to this point, every step of the Claisen condensation is reversible. The next step, deprotonation to form a new stable enolate, drives the reaction forward. To isolate the β-keto ester (or its enol form) requires neutralization or an acid work-up.
As with the aldol reaction, the first step is deprotonation to give the nucleophilic enolate. Deprotonation of an ester is harder than a ketone or aldehyde. The respective pKa values are roughly 25 for the ester and 20 for a ketone, showing that the α-protons of the ketone are more acidic. Conversely, this means the enolate of an ester is more reactive than the enolate of a ketone. It can be argued that the second oxygen attached to the carbonyl group destabilizes the enolate.

Esters are less acidic than ketones. This means they are harder to deprotonate (the α-proton is harder to remove) but that the resulting enolates are more reactive. The difference in reactivity can be explained by the delocalization of the electrons on the second oxygen atom.
Deprotonation with a relatively strong base, such as an alkoxide, leads to an equilibrium favoring the ester but provides sufficient enolate that reaction can proceed by a nucleophilic addition. The enolate attacks a second molecule of ester and leads to the tetrahedral intermediate. The tetrahedral intermediate is different to that found in the aldol addition as it has a leaving group attached, which allows collapse to reform the carbonyl group (see HERE). This results in the formation of a β-keto ester.
The problem with the reaction is that the tetrahedral intermediate has two leaving groups. Both the alkoxide or the original enolate can be kicked out. In other words, the reaction is reversible. The equilibrium favors the starting materials not the β-keto ester.

The first steps of the Claisen Condensation are in equilibrium and they favor the starting materials. Deprotonation of the β-keto ester is irreversible. This gives a delocalized anion, and it is the stability of this intermediate that drives the reaction forward. To isolate product you need to neutralize the second enolate.
The reversibility of the initial addition is highlighted by the failure of disubstituted enolates to give the desired product. The addition occurs but there are no α-protons between the two carbonyl groups and the final deprotonation is not possible. Instead, the base can add to the carbonyl group and the tetrahedral intermediate collapse to give an ester and the enolate. The enolate will deprotonate any methanol due to its higher basicity. Overall entropy and the weak base mean the starting esters are favored over product.

The equilibrium prevents the formation of disubstituted β-keto esters as there is no longer an α-proton to be irreversibly deprotonated. This can be overcome if a very strong base is used to drive the initial deprotonation forward.
The reaction of disubstituted enolates can be forced to completion by the use of a strong base that makes the initial deprotonation essentially irreversible.
Fortunately, if there is only a single substituent on the enolate, this is not an issue as the central α-protons of the β-keto ester are more acidic than the starting materials. A second deprotonation occurs to give a new delocalized enolate. Formation of this stabilized anion is favored and this step is irreversible under the reaction conditions. The pKa of the α protons of a β-keto ester are approximately 10 (acetylacetone) while the pKaH of conjugate acid of the base is 16 (methanol). This difference is sufficient to drive the reaction forward, and the desired product is formed. If you want to isolate the product, the delocalized enolate must be neutralized in an acidic work-up.
This irreversible step marks a second difference to the aldol reaction. The aldol addition (and condensation) can use a sub-stoichiometric (catalytic) quantity of base. The Claisen condensation is not catalytic in base, requiring at least one equivalent. The choice if base is also important as you are dealing with an ester. The wrong base can lead to transesterification and the formation of a mixture of products. The alkoxide base should match the ester.
Transesterification can be an issue in the reaction of any ester with an alkoxide base. The problem is that the base can act as a nucleophile and change the ester through an example of acyl substitution. This can be avoided by using very bulky alkoxides or, more normally, by matching the base to the alkoxy substituent. In the latter case, the transesterification reaction still occurs but as the starting material and product are the same no one notices.
Like the aldol reaction, the crossed, or mixed, Claisen condensation increases the scope of the reaction by allowing two different molecules to be joined together. The key is controlling the order of addition, which molecule behaves as the nucleophile and which behaves as the electrophile. Otherwise, you will form a mixture of compounds. The easiest way to control the addition is to ensure only one molecule can form an enolate:

The mixed Claisen condensation can be achieved when only one ester has α-protons and so is the only component that can form the appropriate nucleophile. In this example, only ethyl acetate will form an enolate and this will attack the other ester to form the tetrahedral intermediate. Ideally, one coupling partner should be more electrophilic than the other to avoid self-condensation.
The mixed Claisen condensation follows the same reaction mechanism as before, the key difference in this example is that only one of the ethyl esters has α-protons. This is the only starting material that can be deprotonated and form the appropriate enolate. The nucleophilic enolate attacks the other ester. This is probably slighly more electrophilic due to the electron withdrawing aromatic ring (the final product will also be more favorable due to delocalization across the whole molecule). Addition gives the tetrahedral intermediate. This collapses to regenerate the carbonyl group in the form of a ketone and to kick out the leaving group, the alkoxide. All of these steps are reversible and it is necessary for the base to deprotonate the β-keto ester creating a fully conjugated molecule and driving the reaction forward.
Crossed Claisen condensations can occur between a ketone and an ester. The ketone is the nucleophile for the reaction to be a Claisen condensation. Ketones are more acidic than esters, and will be more readily deprotonated; remember the additional oxygen on the ester hinders enolate formation (see above) so ethyl acetate has a pKa ~25 while that of acetone is ~20. But, as before, the best yields are achieved if only the ketone can be deprotonated and the ester has no α-protons.

Another example of a mixed or crossed Claisen condensation. This involves the reaction of a ketone with α protons and a carbonate. The carbonate is a more electrophilic version of an ester as it has an additional electron withdrawing group on it. This is a nice route to β-keto esters.
To avoid self-condensation of the ketone in an aldol reaction, a better electrophile is used as the second starting material. In the example above this is a carbonate. Carbonates are better electrophiles than normal esters and even ketones due to the second alkoxy group. Normally esters have a similar or reduced electrophilicity to ketones as a lone pair of electrons on the alkoxy oxygen is delocalized and reduces the partial positive charge on the carbon. But a carbonate has two alkoxy groups and the second acts in a purely inductive fashion increasing the electrophilicity.
The Claisen condensation is summarized in the diagram below:

A generalization of the Claisen condensation and its mechanism.
The Dieckmann Condensation (or intramolecular Claisen condensation)
If the nucleophile and electrophile of the Claisen condensation are joined together, the reaction is a cyclization and is known as the Dieckmann condensation. The intramolecular Claisen condensation is a good method for the formation of five and six-membered rings. It also highlights my dislike of learning organic chemistry through named reactions; it's the same reaction, with the same mechanism, all that has been changed is that the purple and green circles in the diagram above are linked/merged into a horrible colored line (some kind of brown if I remember correctly).

The Dieckmann condensation is the intramolecular variant of the Claisen condensation. The mechanism is identical, with the same limitations, the only difference is that the two esters are now connected and the product is a cyclic molecule.
Like the Claisen reaction, it is possible to use either ketones or esters in the Dieckmann condensation. The reaction below shows such an example. There are three positions that could be deprotonated to form an enolate and yet only one reacts. There are several reasons for this. Firstly, the ketone is more acidic than the ester, so it is more likely to form an enolate (see above). Secondly, only one position leads to a fused bicyclic ring (two rings sharing two atoms and a bond). This is important as it is the only position that permits the irreversible deprotonation to give the delocalized anion. Attack from the other side of the ketone would lead to a bridged ring system (the two rings share two atoms but multiple bonds).

An example of the Dieckmann condensation between a ketone and an ester.
The Mannich Reaction
The Mannich reaction replaces the electrophilic carbonyl group with an iminium ion, the charged nitrogen equivalent. This means the product is a β-amino ketone rather than an alcohol otherwise the mechanism is the same. The classic Mannich reaction involves the reaction of formaldehyde with a secondary amine all in the presence of a ketone and an example is shown below:

The ‘classic’ Mannich reaction involves the addition of a ketone to formaldehyde and a secondary amine in the prescence of acid. Formaldehyde is used as it is more electrophilic than a ketone and this ensures the amine condenses with it prior to enolate addition.
Formaldehyde is the most commonly used aldehyde in the Mannich reaction as it is highly electrophilic. This is necessary for the components of the reaction to couple in the correct order. Formaldehyde condenses with the amine to form an iminium ion (see HERE for a reminder). Formaldehyde is more reactive than the ketone, it is more reactive than most aldehydes! Ketones are less electrophilic than aldehydes as the hydrogen on the carbonyl group has been replaced by an electron donating and bulky substituent. This reduces the partial positive charge on the carbon and hinders the approach of the nucleophile. Formaldehyde has no alkyl groups and only hydrogen atoms on the carbonyl carbon. Nothing reduces its reactivity.
The first step of the Mannich reaction is the condensation of amine and aldehyde as shown below. The first step is probably protonation of formaldehyde (protonation is necessary, the amine will attack formaldehyde directly but the reactions are performed under acidic conditions). The amine attacks the oxonium species to give a tetrahedral intermediate. Proton transfer leads to a hemiaminal and then oxonium species. It is the latter step that requires the acid catalyst. Collapse of the tetrahedral intermediate leads to dehydration and formation of an iminium ion. Many of these iminium salts are surprisingly stable.

The highly reactive aldehyde, formaldehyde, reacts with dimethylamine to give an iminium ion. The mechanism is a condensation reaction. There are a number of variants of this mechanism. The amine will attack formaldehyde prior to protonation but there must be an acid present to form the leaving group for the final step. Sometimes you will see the proton transfers as intramolecular steps and sometimes you’ll see water acting as the base to transfer the proton. All of these mechanisms are acceptable. Proton transfers are rapid and equilibriums so all are probably occurring.
The iminium ion is highly electrophilic and will react with an appropriate nucleophile. Under that acidic conditions keto-enol tautomerization creates a small amount of nucleophilic enol from the ketone. The two reacts add to give the protonated product. Deprotonation leads to the final product and regenerates the proton source.

Acid-catalyzed keto-enol tautomerization leads to the formation of the nucleophilic enol species. This attacks the highly electrophilic iminium ion in an aldol-like reaction to give an amine. A final proton transfer regenerates the acid catalyst.
The similarities between this and the aldol reaction should be readily apparent. The difference is in the electrophile; instead of an aldehyde or ketone there is the more electrophilic, charged iminium species. This is the nitrogen equivalent of an oxonium ion.
It is possible to build more complex molecules using the Mannich reaction, either by pre-making the iminium ion prior to the addition of the nucleophile or by carefully selecting the aldehyde and ketones used in the reaction to ensure that the correct iminium species is formed. As with mixed or crossed aldol reactions, the easiest way to achieve this is by using a ketone with α-protons that will form the nucleophile, and an aldehyde with no α-protons. The latter cannot form a nucleophilic enol and is normally more electrophilic than a ketone so will preferentially react with the amine to form the appropriate iminium species. An example of the latter strategy is taken from the work of Benjamin List, a Nobel prize winner, and is shown below:

In this example of the Mannich reaction there is a condensation between an electron rich aromatic amine (an aniline called p-anisidine) and an electron poor aromatic aldehyde. The proline acts as a catalyst and converts acetone to an enamine, an enol-like reactant that behaves as a nucleophile. The Mannich reaction is followed by hydrolysis of an iminium species to give the product.
While the reaction above is an example of a Mannich reaction, it proceeds through a different mechanism to that discussed in this summary. The proline catalyst, which is used to control the enantioselectivity of the reaction, also converts acetone into a nucleophilic enamine rather than an enol. I haven’t covered this chemistry yet so will not include the mechanism here. I will explain it in a subsequent summary. Bottomline is that the mechanism is the same except a nitrogen atom replaces the oxygen of the enol.
The Mannich reaction is summarized in the diagram below:

A generalization of the Mannich reaction and its mechanism.
The Henry Reaction or Nitro-Aldol Reaction
Carbonyl groups are not the only functional group that undergo tautomerization to create a nucleophilic species. This means that aldol-like chemistry is not restricted to aldehydes, ketones and esters. The nitro group forms an enol like species and acts as a nucleophile in the Henry reaction or nitro-aldol reaction. This is an attractive reaction as it introduces a nitrogen into the molecule. An example is shown below:

An example of the Henry or nitro-aldol reaction. This the addition of a nitroalkane to an aldehyde to give, initially at least, a β-nitro alcohol.
Nitroalkanes are in equilibrium with a tautomeric form known as the aci-nitro form. Normally, the addition of base promotes the formation of the nitro-stablized anion or nitronate. The similarity between these species and carbonyl compounds should be obvious.

The enolization of a nitroalkane is analogous to that of an aldehyde (or ketone or ester). The resulting species is a good nucleophile.
Both aldehydes and nitro groups can tautomerize, with the shift of a proton from carbon to oxygen and the formation of a new double bond. Both of the new species are nucleophilc through the carbon atom with the electrons flowing from an oxygen lone pair to form a double bond and allow the carbon to attack a suitable electrophile. This is even clearer if you look at the deprotonated version, the enolate or nitronate. In both cases, the anion is delocalized with the negative charge on either the oxygen or carbon.
Below is a second example and the mechanism of the Henry reaction. Nitromethane can be deprotonated to give the nitro-stabilized anion. The nucleophilic anion attacks the ketone in an example of nucleophilic addition to the carbonyl group as described HERE. The resulting alkoxide deprotonates more of the nitroalkane or any water present to give the product. You should see this is the same as the aldol reaction; deprotonation leads to a delocalized anion that acts as a nucleophile and attacks a carbonyl group. Proton transfer gives the product.

An example of the Henry reaction or nitro-aldol reaction. The mechanism is shown. This starts with deprotonation to give the nucleophilic nitronate that attacks the ketone. The resulting alkoxide is sufficiently basic to deprotonate nitromethane to give the product and form more nucleophile.
You might think that the aldol reaction would compete with the Henry reaction, surely the base could deprotonate the ketone and give an enolate? This is very rarely a problem. The nitroalkane is considerably more acidic than a ketone with the former having a pKa ~10 while a ketone has a pKa >20. The nitroalkane is preferentially deprotonated to give the more stable anion. This means the Henry reaction can be promoted by weaker bases than hydroxide and you will often see carboxylate salts such as sodium acetate or amines used as the base.
The increased acidity of α-protons can be problematic as it means the product is readily deprotonated leading to dehydration and formation of a nitroalkene through an E1cB mechanism. With aromatic aldehydes this is almost impossible to stop. An example is shown below:

The acidity of the α-protons of a nitro group make addition followed by condensation a common reaction pathway. In the case of aromatic aldehydes the condensation is hard to prevent. The reaction starts with the Henry addition to give the β-nitro alcohol. A second deprotonation gives a nitro-stabilized anion that undergoes elimination, kicking out a hydroxide anion.
The mechanism of the reaction starts the same as before with the first step being deprotonation to give an nitronate. Nucleophilic addition to the aldehyde generates a β-nitro alkoxide. The alkoxide is sufficiently basic to deprotonate more nitroethane or the acetic acid generated earlier in the reaction. As there is a second α-proton, there is a second deprotonation to give a nitro-stabilized anion. This eliminates the hydroxide anion to give a nitroalkene. The elimination is almost certainly E1cB due to the poor leaving group ability of the hydroxide anion.
The nitro group is a useful functional group that can be reduced to an amine or oxidized to a carbonyl group (the Nef reaction). Such reactions may be covered in a different summary (but don’t hold you breathe as it isn’t on my plans at the moment!).
The Henry reaction or nitro-aldol and the subsequent dehydration is given below:
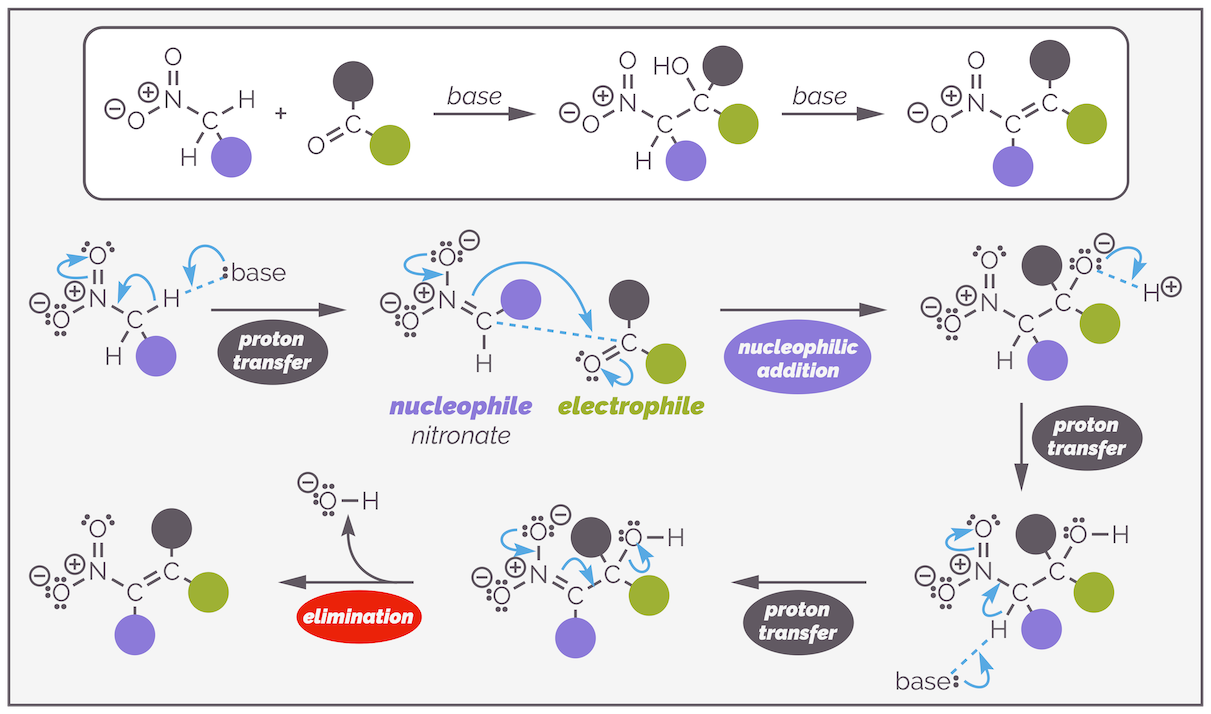
A generalization of the Henry or nitro-aldol reaction and subsequent dehydration leading to a nitroalkene.
Conclusions
The general mechanism behind the aldol reaction can be applied to a range of useful transformations involving a variety of different functional groups. Generally speaking, one component can form a nucleophile by either tautomerization or deprotonation to a delocalized anion. The other coupling partner contains an electrophilic carbonyl group (or equivalent, as in the case of the Mannich reaction). The two reactants add to form a new C–C bond by nucleophilic addition. In many examples, a subsequent dehydration step leads to a C=C double bond.
The Claisen condensation involves the formation of a nucleophilic enolate that then attacks an ester (or equivalent functional group) leading to the formation of a β-keto carbonyl group. Two carbonyl groups are retained within the product (although enolization may hide one) due to the presence of a leaving group on the electrophile. The Dieckmann condensation is the intramolecular variant of the Claisen condensation. It is follows the same mechanism but the leads to cyclization as both the nucleophile and electrophile are connected.
The Mannich reaction replaces the electrophilic carbonyl group of the aldol reaction with an iminium ion. Otherwise the reaction is the same, with an enol or enolate attacking the polarized π bond to give a β-amino carbonyl molecule.
Finally, for this summary, is the Henry reaction or nitro-aldol. This replaces the nucleophilic aldehyde, ketone or ester with a nitroalkane. But, once again, the mechanism is the same deprotonation to give a delocalized anion forms a nucleophile that attacks an aldehyde or ketone to give a β-nitro alcohol.
Those of you wanting to take this further can probably already see that you could mix and match these reactions. There is no reason that you couldn't use a nitroalkane to attack an iminium species and form a β-nitro amine for instances.
The idea that a single mechanism effectively explains a variety of reactions (Claisen addition, Dieckmann reaction, Mannich reaction and the Henry reaction amongst others) shows the power of arrow pushing (and yes, I’m well aware that these mechanism are far from ‘true’ but their predictive power is undeniable). It also reveals why organic chemistry is so versatile; by applying a single idea, a host of different molecules can be constructed. It also highlights one of my bugbears about the teaching of organic chemistry in that you can find many books and websites that list ‘named' reactions. They separate out and classify reactions almost alphabetically, ignoring the connections between molecules. It is no wonder that many students are scared of organic chemistry. They are given lists of reactions to learn and ignore the very limited set of principles that cover all the reactions.
In later summaries, I will expand upon the simple principles already covered to allow you to gain more control over your reactions and you will start to look at regio- and stereoselectivity.