Lithium enolates & enolate equivalents
Introduction
The previous summaries have covered the basic concepts behind the formation of enols and enolates as well as their most common reactions. These reactions are often grouped as aldol-like reactions as they share a common mechanism. I have skirted around the subject of selectivity, and often chose examples that could only give a single product. Crossed or mixed aldol reactions, where two different carbonyl-containing compounds react are very useful if you can control the selectivity of the reaction; which substrate will be the nucleophile and which the electrophile? Can you control the regioselectivity of deprotonation? Can you control the stereoselectivity? This summary will look at ways of controlling enolate formation (and the effect this can have on the aldol reaction).

The problem with the crossed or mixed aldol reaction is controlling the selectivity. How do you ensure only one substrate acts as the nucleophile? How do you control the regioselectivity of deprotonation and how do you control the stereoselectivity of the resulting β-hydroxy ketone?
One of the most common methods to control the selectivity of a crossed aldol reaction is to form the lithium enolate by reacting one of the substrates with a strong base at low temperature prior to adding the electrophile. Alternatively, you can prepare an enolate equivalent, reactive functionality that behaves like an enolate but offers distinct advantages (and some disadvantages). In this summary, I'll provide more details about these two approaches.
Lithium enolates
To prevent the self-condensation of a carbonyl-containing compound during an aldol reaction, as outlined in the previous summary HERE, you must ensure that all of the carbonyl compound is converted to an enolate prior to any nucleophilic addition occurring. This leaves you with a nucleophile that has no electrophile to attack, no electrophile, no self-condensation. Reaction with a strong base, often LDA (lithium diisopropylamide), gives the lithium enolate.
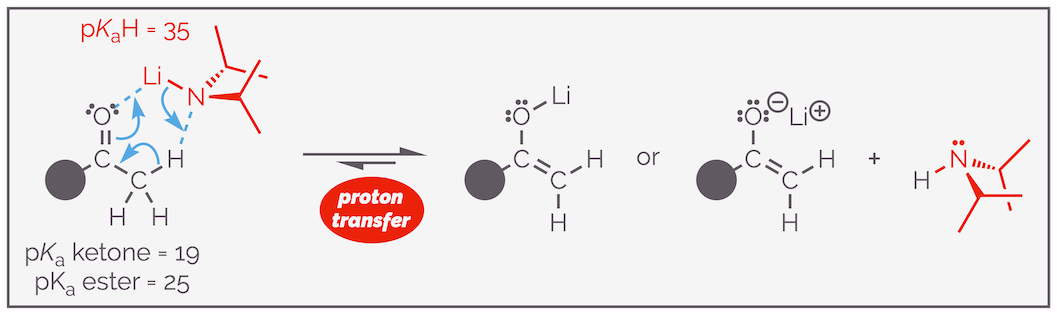
Deprotonation of a methyl ketone with LDA (lithium diisopropylamide) leads to the formation of the lithium enolate. The large difference in pKa means the reaction is essentially irreversible. There are two common representations of lithium enolates, one has a O–Li bond, the other depicts the molecule as ionic (remember that the majority of bonds are somewhere in between the two extremes. Also the structure of lithium enolates is far more complex than indicated in organic textbooks with these species being dimers, tetramers or even larger complexes. There was a fascinating Seebach paper in the late 80s on the subject).
Lithium enolates are sufficiently stable at –78 °C that they will not react or add to a carbonyl group. LDA is a strong enough base to rapidly deprotonate the α proton even at such low temperatures. Quick inspection of the difference in the pKa between the conjugate acid of LDA and that of ketones, esters, or amides (~ 10 units) indicates that the reaction is effectively irreversible. The enolate can be made in the presence of the carbonyl without undesired reaction occuring. The pre-formation of lithium enolates prior to the addition of the electrophile permits crossed or mixed aldol reactions even with highly enolizable aldehyde substrates. A limitation of the LDA chemistry is that it cannot be used to form lithium enolates from aldehydes, it participates in nucleophilic attack on the aldehyde.

An example of a mixed aldol reaction. The enolate is formed prior to the addition of the aldehyde. In this example, I’ve used the Ireland model of deprotonation, which involves a cyclic six-membered transition state. The aldol reaction also proceeds through a cyclic transition state with both the enolate and the aldehyde coordinated to the lithium cation (this is probably incorrect and the complex is more, well complex but it gives a use-able explanation of the observed results, which is that the so called syn-aldol is favored).
The next paragraph is far more detail than most undergraduates need but it becomes useful when looking at stereoselectivity Deprotonation to form the enolate is often depicted as proceeding through a cyclic transition state (the Ireland model) and this becomes important if you need to control stereochemistry. Alternatively, the geometry of the enolate can also be explained by looking at a simplistic view of the molecular orbitals. Deprotonation to give an enolate occurs when the σ C–H bond is perpendicular to the carbonyl π* antibonding orbital. Two conformations allow this, one places the methyl group cis to the phenyl ring while the other has it trans. The latter is favored on steric grounds.

The optimum conformation for the deprotonation of an α-proton and formation of an enolate has the maximum overlap of the C–H σ orbital and the C=O π* antibonding orbital. There are two conformations that permit this overlap. One has an unfavorable interaction between the methyl group and the phenyl group.
As LDA leads to rapid and irreversible deprotonation at low temperatures, it is used in regioselective enolate formation of unsymmetrical ketones. It favors the kinetic enolate and the preferential removal of one proton can be rationalized through three arguments. Firstly, LDA is a bulky base and will remove the most accessible proton, the proton on the least substituted carbon, simply to avoid steric interactions. Secondly, the protons on the less substituted carbon are more acidic as the carbanion resonance structure has fewer destabilizing inductive interactions. Thirdly, there are more protons on this carbon so there is more chance of them being removed. All this is shown in another example of the crossed or mixed aldol reaction below:

An example of a crossed aldol reaction that starts with the regioselective formation of the kinetic enolate. LDA is a bulky base and, at low temperature, will preferentially remove the least sterically hindered proton. This is the proton on the least substituted carbon. At low temperature, this reaction is essentially irreversible. This example shows the methyl group causing the hindrance. After enolate formation, an electrophilic aldehyde is added. The reaction proceeds through a cyclic transition state (or at least it can be simplified to this transition state) with the aldehyde approaching from the least sterically demanding side of the enolate (away from the methyl group). The relative stereochemistry can be predicted from the cyclic transition state.
The use of LDA to form lithium enolates at low temperatures facilitates the crossed or mixed aldol reaction of carboxylic acid derivatives as well. The reaction proceeds along the same pathway as above; the enolate is formed in isolation. The electrophile is then added and the two allowed to react (normally by warming the reaction mixture up). Finally, a weakly acidic work-up protonates the alkoxide intermediate and gives you the product.

The use of LDA in the crossed or mixed aldol reaction allows ketones, amides and esters to be transformed into nucleophiles prior to the addition of the electrophile.
Silyl enol ethers
Silyl enol ethers are useful enolate equivalents as they are easy to form, are more stable than lithium enolates and yet, will still participate in nucleophilic additions such as the aldol reaction. The latter characteristic leads to their description as enolate equivalents, a class of compound that are not an enolate but behave as if they were. There are numerous methods to form silyl enol ethers and this permits control over both the regio- and stereo-selectivity. There are two common methods for their formation. The first involves trimethylsilyl chloride (TMS–Cl; (CH3)3SiCl, Me3SiCl) and a weak base, such as triethylamine. It is shown below:

Silyl enol ethers can be synthesized by the reaction of a carbonyl containing compound, trimethylsilyl chloride and a weak base, such as triethylamine. There are two possible mechanisms for the reaction. Both are driven by the strength of the Si–O bond. The first involves formation of an oxonium ion. The positively charged carbonyl group is highly polarized and pulls electrons towards it. This increases the acidity of the α-protons and means they are readily deprotonated even with a weak base. Alternatively, the silyl chloride reacts with the minor component of the keto-enol equilibrium to give a protonated oxonium ion. This is readily deprotonated to give the silyl enol ether.
There are two proposed mechanisms for this reaction. It is unimportant which is in operation as both will explain how you control regio- and stereo-selectivity. Both mechanisms rely on the strength of the Si–O bond to draw the reaction forward. In the top reaction mechanism, the hard silicon atom reacts with the hard oxygen atom to give an oxonium ion. This charged species increases the acidity of the α-protons dramatically, and now an elimination reaction is possible. All that is required is a weak base and the C–H bond to be perpendicular to the π* antibonding orbital of the C=O group. The alternative pathway sees the reaction proceeding through the enol tautomer (see HERE). Here the base simply mops up the proton that is lost from the oxonium ion.
The second method involves forming a lithium enolate with LDA, or similar strong base, and trapping this with trimethylsilyl chloride. This is one of the few examples undergraduates encounter of enolates reacting at oxygen rather than the carbon atom. Silyl reagents are hard and react with the harder center of the enolate, the oxygen atom. The resulting bond is very strong.
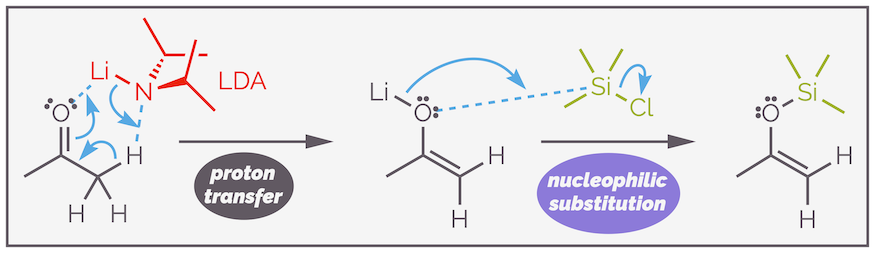
Silyl enol ethers can also be formed by trapping lithium enolates.
The two different methods are very different and permit the regioselective formation of either the thermodynamic or kinetic silyl enol ether. Use of a weak base at higher temperatures favors the thermodynamic product.

Formation of the thermodynamic silyl enol ether is favored when a weak base is used. There are two potential mechanisms; one involves elimination of a proton from a cationic intermediate. The more substituted enol ether is favored as the positive charge is stabilized by the inductive effect of an additional substituent. The second mechanism involves the silylation of the more stable, more substituted enol and then deprotonation of the resulting oxonium species.
There are two common explanations for the observed regioselectivity. Triethylamine is a weak base and cannot deprotonate a ketone. Instead the mechanism proceeds by silylation first. The oxygen attacks the silyl reagent to give an oxonium species. The α-protons of this cationic species are more acidic and can be removed by triethylamine. When deprotonation occurs the cation is effectively transferred from the oxygen to the nitrogen. In the transition state, the positive charge is spread over all the atoms of the newly forming silyl enol ether, the hydrogen atom and the nitrogen. There are two protons that could be removed. One is a secondary proton and the build up of partial positive charge is similar to a secondary cation. The other is more substituted. The build of partial charge is closer to that of a tertiary carbocation. It is more stable due to the inductive effect of the methyl group. This transition state is favored, it is lower in energy. This is the favored, thermodynamic product.
The alternative argument postulates that the reaction proceeds through the more stable, more substituted enol (HERE). Using a weak base at higher temperatures allows the equilibrium to be established and the reaction will favor formation of the more stable silyl enol ether.
The kinetic silyl enol ether is formed if a strong bulky base like LDA is used to form the lithium enolate at low temperature prior to trapping with a silyl reagent.

The regioselective formation of the kinetic silyl enol ether. The reaction is conducted with a strong bulky base at low temperature. The bulky base selectively deprotonates the more accessible, less hindered proton. This forms the least substituted lithium enolate, which is then trapped with a silyl reagent to give the less substituted kinetic enolate.
Using a strong, bulky base at low temperature results in the removal of the least substituted α-proton. This is the more accessible proton so there is less steric interactions slowing the reaction. It is also the most acidic proton and will be removed quicker; the resulting anion is more stable as fewer alkyl groups (substituents) are inductively pushing electron density onto this carbon. This means it will be more acidic.
Silyl enol ethers are less reactive than enolates. Their reactivity is similar to that of an electron rich alkene. The lone pair of electrons on the oxygen can delocalize onto the C=C double bond. This means they react with the same reagents that double bonds react with. A typical example is bromination as shown below:

Silyl enol ethers react with halides. The C=C double bond is electron rich due to delocalization of the oxygen lone pair. It acts as a nucleophile and will attack reactive electrophiles. The silyl group is readily removed from the resulting oxonium ion (or the carbocation resonance structure) to give the neutral product.
The silyl enol ether is a nucleophile, the lone pair of electrons on the oxygen is delocalized and makes the α-carbon (naming still based on the original carbonyl group) electron rich. It will attack suitable electrophiles (sufficiently reactive). The initial step of the mechanism is identical to the reactions of enols and enolates. The anion formed in the substitution step attacks the silyl ether and neutralizes the oxonium species, leaving the brominated ketone.
The reactions of silyl enol ethers require strong electrophiles. A silyl enol ether is not alkylated by a simple haloalkane unless there is a strong Lewis acid present. This activates the haloalkane by forming a carbocation. This is a potent electrophile. This reaction is the same as Friedel-Crafts alkylation (described HERE).

Silyl enol ethers can be alkylated. The reaction requires a Lewis base to activate the weakly electrophilic haloalkane. The Lewis acid rips the halide off to give a carbocation, which is sufficiently electrophilic that it will react with a weakly nucleophilic silyl enol ether.
The electron rich C=C double bond of the weakly nucleophilic silyl enol ether attacks the strongly electrophilic carbocation. This leads to the formation of an oxonium species (shown above) or a delocalized carbocation (resonance structure not shown). The halide anion eliminated from the haloalkene removes the silyl group and neutralizes the charge. This can either be described as a substitution as shown above or an elimination reminiscent of the rearomatization step of the Friedel-Crafts reaction (except a silyl group is removed instead of a proton).
Like alkenes, siyl enol ethers are not sufficiently reactive to attack an aldehyde or ketone unless the carbonyl group is activated. As with the previous reaction, this can be achieved by the addition of a Lewis acid. This is known as the Mukaiyama aldol reaction.

The general outline of the Mukaiyama aldol reaction. This is a reliable method to perform a crossed or mixed aldol reaction with aldehydes.
The purpose of the titanium tetrachloride is to activate the aldehyde. The aldehyde donates a lone pair of electrons to the Lewis acid to form an oxonium species. This cationic species is more polarized than a standard aldehyde so it is more reactive and the weakly nucleophilic silyl enol ether can attack it in an example of nucleophilic addition.

One possible mechanism for the Mukaiyama aldol reaction. The key step that is common to every version of this mechanism is the activation of the aldehyde with a Lewis acid to give a highly electrophilic oxonium species. After that, whether the silicon is transferred inter- or intra-molecularly or at all (there is an argument that the titanium is coordinated between the two oxygen atoms although bonds strengths would suggest the system would favor a Si–O bond over an Ti–O bond), is up for discussion. Ultimately, activation allows the aldol reaction to occur.
The result of the addition is a new oxonium species and an alkoxide. First the trimethylsilyl group is removed by nucleophilic substitution with a chloride ion. This resulting silylating reagent is attacked by the titanium alkoxide to form a new silyl enol ether. This is the stepwise explanation of the reaction and, as such, should be reasonably easy to follow. It is possible that the steps occur in a different order or involve intramolecular silyl transfer. Ultimately, it is not important. All that is important is that the aldehyde must be activated prior to reaction.
The Mukaiyama aldol reaction is one of the best methods for the crossed or mixed aldol reaction of aldehydes. The problem with the crossed aldol reaction of aldehydes is the increased reactivity of aldehydes. This leads to self-condensation being a competitive side reaction. The formation of a silyl enol ether under mild conditions with a weak base, such as triethylamine, avoids self condensation, without the Lewis acid there is no nucleophilic attack. An example is shown below:

An example of the crossed or mixed aldol reaction of two aldehydes. The partial mechanism on the bottom of the diagram shows that formation of the silyl enol ether prevents self-condensation. An enolate is never formed and the nucleophilic silyl enol ether will only react with an activated aldehyde.
Formation of the silyl enol ether does not proceed through an enolate. There is no reactive nucleophile formed in the presence of a good electrophile (unreacted aldehyde). Instead the nucleophile is formed under mild conditions and only reacts when an activated aldehyde, one that has complexed the Lewis acid is added (the more observant amongst you will have noticed that there is an oxonium ion formed during the preparation of the silyl enol ether, so it is actually necessary to lower the temperature to prevent self-condensation).
Similar chemistry can be achieved with esters. The second oxygen means these are no longer called silyl enol ethers but are instead silyl ketene acetals. Otherwise the chemistry is the same (they are slightly more reactive but still require the electrophile to be activated).

An example of the formation of a silyl ketene acetal and its aldol reaction with an aldehyde.
Silyl enol ethers and silyl ketene acetals are weaker nucleophiles than enolates but they are more reactive than alkenes. Their advantage lies in their relative stability that allows them to be isolated prior to reaction. Silyl enol ethers can be made under two sets of conditions: you can mix the carbonyl-containing compound with a weak base and trimethylsilyl chloride (TMS-Cl) at room temperature. This normally gives the thermodynamically more stable silyl enol ether. Alternatively, you can perform the reaction at low temperature with a strong, bulky base, such as lithium diisopropylamide (LDA), to give an enolate that is then trapped by the addition of TMS-Cl. Now the kinetic silyl enol ether is preferred. This means you now have a means of controlling regioselectivity prior to reaction.
Boron enolates
Boron enolates have been extensively used in the aldol reaction. Like silyl enol ethers they provide an isolatable enolate equivalent, a compound that behaves like an enolate (experimentally, you would rarely actually isolate the boron enolate, they are moisture sensitive, instead you prepare them as needed and then add the aldehyde/electrophile). Selection of the correct boron reagent and reaction conditions allows control of the geometry of the enolate. This is important as it can have a strong influence on the stereochemical outcome of boron aldol reactions. As boron is in Group 13 of the periodic table, it can act as a Lewis acid. This means that boron enolates don't require the addition of external Lewis acids like the reactions of silyl enol ethers.
Preparation of boron enolates mirrors the formation of the thermodynamic silyl enol ether and uses a weak base. If the boron reagent is small due to small alkyl substituents, it favors the Z-enolate, with the methyl group of the ketone on the same side as the oxygen atom. The simple mechanism has the Lewis acidic boron forming an oxonium ion prior to deprotonation. This is necessary to increase the acidity of the α-protons as shown by the considerable decrease in pKa.

Formation of the Z-boron enolate is achieved with a small boron reagent. The mechanism involves formation of an oxonium ion prior to deprotonation. The methyl group of the enolate avoids interactions with the bulky phenyl group in the transition state.
The mechanism is undoubtedly more complex with the leaving group on the boron reagent being important. A good leaving group, such as a triflate fully dissociates before the deprotonation. This makes the oxonium species smaller, less sterically bulky and favors the Z-enolate. Poor leaving groups remain associated with the boron in a zwitterionic intermediate that is far bulkier. Also note that I haven't drawn the important transition states (this is meant to be a simple summary!). The boron can either be cis or trans to the alkyl group being deprotonated and the elimination occurs when the proton is perpendicular to the carbonyl (by the time I've written this I should have just drawn it!).
A bulkier boron reagent with a relatively poor leaving group, such as a chloride, favors the E-enolate. The intermediate oxonium ion is bigger, more bulky and forces the methyl group onto the opposite face.

The opposite geometry (stereochemistry) of the boron enolate can be formed using a bulkier boron reagent with a poor leaving group. The mechanism is the same (ignoring whether the halide dissociates or not), it is only configuration of the transition state that changes).
Boron enolates are excellent in the aldol reaction. Boron is Group 13, it has an empty 2p atomic orbital, and acts as a Lewis acid. Not only do you not have to add an external Lewis acid, but, the reaction is intramolecularized. The boron coordinates the aldehyde and now the nucleophile and electrophile are in the same molecule. The transition state is an organized, six-membered chair-like transition state (in fact, the B–O bond lengths are shorter than with other metal enolates, and the transition state is tighter. This increases control by further destabilizing the disfavored transition states). This allows control of stereochemistry. The coordination of the boron and aldehyde also makes the nucleophile more nucleophilic (the enolate is activated) and the electrophile more electrophilic (polarizes the carbonyl group). Below shows the reaction of the Z-enolate:

Selective formation of the Z-boron enolate followed by crossed or mixed aldol reaction. The reaction forms a new C–C bond and controls the relative stereochemistry of the two new stereocenters. The key to the selectivity is the six-membered chair-like transition state.
The Z-enolate favors formation of the syn diastereomer in a diastereoselective or stereoselective reaction. The reaction is selective not specific as two possible diastereomers are possible by the same reaction mechanism but one is favored. In a stereospecific reaction, the mechanism only permits a single stereoisomer to form (think SN2). The stereoselectivity is controlled by a chair-like transition state that can be drawn to resemble the conformation of cyclohexane (HERE; it isn't identical to cyclohexane, there are four sp2 hybridized atoms in the ring but this approximation works. This is one of the reasons organic lecturers are obsessed with the conformation of cyclohexane). The position of the enolate substituents are fixed by the geometry of the double bond (in my experience, this is the bit some students struggle with, they want to place all the substituents pseudo-equatorial even when this is not possible). The only choice in the system (as long as we ignore absolute stereochemistry) is the orientation of the aldehyde. The substituent of the aldehyde (R2 will adopt the pseudo-equatorial position to minimize 1,3-diaxial interactions with the substituent of the enolate (R1). As always, this is a simplification that works well for simple achiral aldehydes, the situation becomes more complex if there is an α-stereocenter and then you have to worry about syn-pentane interactions but that is a far more advanced topic than this summary deals with.
If the geometry of the enolate changes so the favored diastereomer changes; the E-enolate favors the anti diastereomer. There is no change in the reaction. There is no change in the explanation, you have simply altered the position of one substituent. This is shown below:

The E-enolate favors the formation of the anti diastereomer. The explanation is the same as before; the reaction proceeds through a chair-like transition state and the orientation of the aldehyde is such to minimize 1,3-diaxial interactions. The position of all the other groups is fixed. The fact the geometry of the enolate is different means the favored diastereomer is different.
Boron enolates have been used extensively in the synthesis of Natural products and biologically important compounds. It is possible to add chiral ligands to the boron (L in the diagrams above), and this permits control of the absolute stereochemistry as well as the relative stereochemistry. This is powerful and useful chemistry.
Conclusion
Enolate chemistry allows carbonyl groups, such as aldehydes, ketones, esters, and amides, to acts as nucleophiles and functionalize the α-position. At undergraduate, the basic concepts are covered but the examples given are frequently carefully picked, they are very specific and only work for a handful of molecules under particular conditions. In a real lab situation, chemists frequently use stable enolates or enolate equivalents (molecules that behave as if they were an enolate).
The most common examples are lithium enolates, silyl enol ethers or boron enolates (and enamines, which I’ll cover in a different summary). Lithium enolates are made with strong bases and are relatively stable at -78 °C. This means they can be formed without reacting with an electrophile, and this allows crossed or mixed aldol reactions. The use of a strong base at low temperature means you can control the regioselectivity of deprotonation of an unsymmetrical ketone so that it favors the formation of the kinetic enolate.
Silyl enol ethers are more stable than enolates yet are still reactive. Formation of a silyl enol ether can be achieved under either thermodynamic or kinetic control. The thermodynamic silyl enol ether is formed with a weak base at elevated temperature while the kinetic silyl enol ether is prepared by trapping the lithium enolate with a silyl chloride. This allows chemists to control the regiochemistry at will. Silyl enol ethers react with strong electrophiles directly but if you want to achieve an alkylation or aldol reaction you need to activate the electrophile with a Lewis acid.
Boron enolates are very effective in the aldol reaction. Boron acts as an internal Lewis acid and this intramolecularizes the reaction. Depending on the boron reagent used, it is possible to control the geometry or stereochemistry of enolate formation. This in turn, then controls the relative stereochemistry of the aldol reaction. Boron enolates are very important in stereoselective synthesis and I'm sure I'll be discussing them again at some point.
Hopefully, this summary presents an introduction to controlling selectivity in reactions.
There is one more enolate equivalent that I haven’t covered yet and that is enamines. I'll discuss them in the next summary.