Enamines
Introduction
For the last couple of summaries, I’ve discussed the chemistry of enols, enolates, and enolate equivalents, functionality that behaves like an enolate but is more stable. One group has been missing from this discussion, enamines. Enamines are the nitrogen equivalent of an enol. As nitrogen is less electronegative, they are more reactive than enols while offering numerous advantages. Enamines have formed the cornerstone of organocatalysis, one of the game changers in organic chemistry in the last 20 years. In this summary I am only introducing this versatile functional group and will concentrate on the reaction below:

General enamine reaction
Alkylation of a ketone proceeding through an enamine. The reaction of pyrrolidine and cyclohexanone leads to the formation of the nucleophilic enamine. This can react with suitable electrophiles to give, after hydrolysis, the alkylated product.
Enamine Formation
When a primary amine reacts with a ketone, a condensation or dehydration reaction occurs to give an imine. The mechanism of this reaction was detailed in an earlier summary HERE. Key to this reaction are the two protons on the amine. These allow the formation of water and can be removed from the molecule to give a neutral species. A reminder of the mechanism is below:

Imine formation
The reaction of a ketone and a primary amine leads to the formation of an imine. This is a condensation reaction that proceeds with the elimination of water. Lose of the N–H proton from the charged iminium species (deprotonation) leads to the neutral imine. This is only possible if a primary amine (or ammonia) is the initial nucleophile.
When a secondary amine, an amine with two substituents and only one N–H bond, is used, the final deprotonation (bottom left deprotonation step in the scheme above) is impossible, there is no N–H bond on the iminium ion to remove. It is impossible for a secondary amine to give an imine without cleaving a strong N–C bond. While the condensation proceeds the same as for a primary amine, once the iminium species is formed the mechanism mirrors enolization and the α-proton (C–H) is removed instead. This leads to the formation of an enamine (en = alkene and amine = amine).

Enamine formation
The reaction of a secondary amine and an aldehyde or ketone leads to an enamine. The mechanism starts the same as imine formation. The amine attacks the polarized carbonyl group in an example of nucleophilic addition. There is a series of proton transfers that leads to the formation of an oxonium leaving group. The water is kicked out to form an iminium species. Unlike with imine formation, there is no N–H bond that can be broken to give a neutral species. Instead it is necessary to deprotonate the α-position leading to an enamine, a species that looks like a enol except with a secondary amine replacing the oxygen.
Every step of the mechanism is reversible and enamines, iminium ions and imines are not stable to aqueous acid. All three are readily hydrolysed to give either an aldehyde or ketone. At first glance, this may seem like a disadvantage but it is key to their use as organocatalysts , allowing a sub-stoichiometric (less than one equivalent) quantity of amine to react with multiple equivalents of carbonyl compound.

Reversibility of enamine formation
Just like imine formation, enamine formation is an equilibrium reaction. This means enamines are unstable in the presence of aqueous acid. The mechanism is the reverse of enamine formation. Protonation of the enamine leads to the formation of an iminium cation. Water attacks the charged species leading to a tetrahedral intermediate that resembles an acetal. Protonation of the amine forms a good leaving group that is expelled to give a ketone.
While any secondary amine can be used in enamine formation, it is more common to see cyclic secondary amines. Generally, cyclic secondary amines are more nucleophilic than acyclic amines as the alkyl groups are tied back. Cyclic amines also lead to a more nucleophilic enamine. Again, this is due to the alkyl groups being tied out of the way but there is also an argument that hyperconjugation or σ-conjugation increases the nucleophilicity in cyclic amines. This states that the overlap of the C–H bond with the nitrogen lone pair increases its nucleophilicity. Finally, the common cyclic secondary amines have reasonably high boiling points that means that enamine formation can be performed at a higher temperature making it easier to drive off water and force the equilibrium in favor of the product.
The reaction of non-symmetric ketones occurs with good regioselectivity, favoring the thermodynamic enamine. Every step of the reaction is reversible so it favors the most stable compound. The structure of the thermodynamic enamine isn’t as clear cut as with enols. With cyclic ketones, the thermodynamic product of an enamine is normally the less substituted double bond as this minimizes steric interactions between the substituent on the nitrogen and the double bond substituent. Such an interaction is not possible with enols and this accounts for the reversal in selectivity.
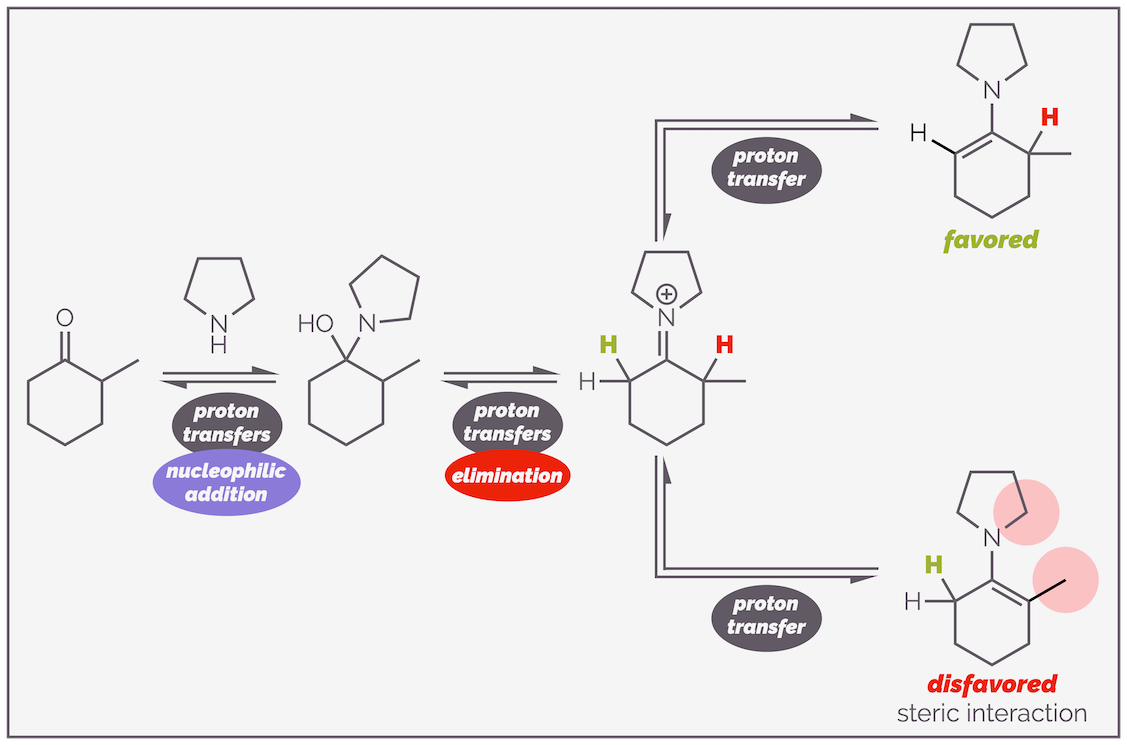
Enamine formation of an unsymmetric ketone leads to the thermodynamically more stable, less substituted enamine. This minimizes the interaction between the alkyl groups attached to the nitrogen and the substituents on the C=C double bond. Each step of the reaction is reversible and this explains why the most stable, least sterically congested compound is favored.
A quick aside:
A common question from students is why doesn’t the amine simply rotate around the N–C=C single bond to alleviate the strain? Placing the pyrrolidine ring perpendicular to the cyclohexene ring would remove the steric interaction as shown in the diagram below.. To answer this you need to think about why enamines are nucleophilic. The lone pair of electrons on the nitrogen is delocalized into the double bond. It can only do this if the appropriate orbital (2p orbital not sp3; remind yourself why HERE) is parallel with the 2p orbitals that overlap to make the π bond. This is shown in the diagram below.

Trying to understand why enamines cannot rotate to minimize steric interactions. The lone pair of electrons on the nitrogen is delocalized into the alkene creating double bond-like character.
Enamines are planar with the three atoms of the enamine all being sp2 hybridized. This means the pi electrons are delocalized and there is double bond-like character to the C–N bond preventing free rotation.
Going back to the thermodynamic enamine, if it is an acyclic ketone, and there is a single substituent at the α-position then the thermodynamic enamine is the more substituted alkene most of the time … but it depends on the substituents on either side of the ketone. Bottom-line, regiochemistry is excellent in certain examples but is not as straightforward as some textbooks suggest.

The thermodynamic enamine of an acyclic ketone is similar to an enol. It normally favors the more substituted double bond as these are more stable.
Enamines are readily formed from the reaction of an aldehyde or ketone with a secondary amine. Once formed, enamines are good nucleophiles.
The Reaction of Enamines
Enamines are good nucleophiles. They are more nucleophilic than enols or silyl enol ethers (HERE) as the nitrogen is less electronegative than an oxygen. It more readily donates its lone pair of electrons to allow delocalization across the enamine. The increased delocalization places enamines between enols and enolates in terms of reactivity.

In enamines, the nitrogen lone pair of electrons os delocalized. Both the nitrogen and the α carbon are nucleophiles. Due to the the reduced electronegativity of nitrogen compared to oxygen, the delocalization is more important, and enamines are more nucleophilic than enols.Make it stand out
Enamines will react with haloalkanes to give the product of α-alkylation. The reaction is best represented as the lone pair of electrons on the nitrogen being shared between the nitrogen and carbon to created a double bond. This forces the enamine to react through the α-carbon atom and attack the haloalkane in a substitution reaction.

The alkylation of an enamine. Enamines are nucleophilic with the electrons of the nitrogen moving to form an iminium cation. As they do this, the electron rich C=C double bond attacks the haloalkane in an example of nucleophilic substitution. The resulting iminium cation is unstable and is normally hydrolysed with weak aqueous acid. This is the reverse of imine formation. It involves nucleophilic addition followed by proton transfers. The amine is eliminated to give an oxonium ion that undergoes a final proton transfer.
The mechanism involves nucleophilic substitution with the enamine attacking the haloalkane and kicking out the leaving group. This results in the formation of an iminium species that is hydrolyzed. The mechanism was discussed above and HERE. Water attacks the highly electrophilic iminium ion. There are a series of proton transfers that create a good leaving group in the form of a quaternary amine. This is eliminated when the oxygen lone pair flows back down to create the carbonyl group. A final deprotonation gives the product.
Care must be taken, only the most reactive haloalkanes participate in α-alkylation. Less reactive haloalkanes react at the nitrogen atom (it is still a good nucleophile), and this leads to formation of the original aldehyde or ketone and a tertiary amine. Once the nitrogen has been alkylated it has no lone pair of electrons to activate the C=C double bond and hydrolysis is the only possible route to a non-charged compound. Activated haloalkanes are normally adjacent to a double bond as shown below:

Reactive haloalkanes will be adjacent to a double bond (or heteroatom). They include allylic halides, benzylic halides and halides α-carbonyl group.
Enamines are good nucleophiles for conjugate additions or Michael additions. Chemoselectivity can be an issue in conjugate additions as there are two electrophilic positions, the conjugated alkene (β or 4 position) and the carbonyl group (2 position). See HERE if you need a quick recap. Enamines are soft nucleophiles as the electrons are delocalized, making them large and diffuse. Soft nucleophiles attack the desired soft position of a Michael acceptor (conjugated enone, enal, or enoate), which is the β-or 4-position (soft likes soft). Even if they do attack the carbonyl group, 1,2-addition is reversible and the enamine is readily eliminated. This allows equilibrium to favor formation of the 1,4-adduct.

A Michael or conjugate addition involving an enamine attacking a enoic acid. The soft nucleophile attacks the soft 4-position of the enoic acid resulting in the eventual formation of a 1,5-dicarbonyl compound. The mechanism involves conjugate (nucleophilic) addition followed by proton transfer and then hydrolysis of the iminium ion.
The mechanism of the reaction is effectively the same as the alkylation reaction. The electrons flow from the enamine nitrogen leading to nucleophilic attack from the α-carbon onto the activated alkene. This gives a zwitterion, a species containing both a positive and negative formal charge. The negative charge is an enolate, formed when the electrons of the enoic acid flowed towards the electronegative oxygen atom. The enolate is protonated to leave the iminium cation. A weak aqueous acid hydrolyses the iminium ion by nucleophilic addition of water followed by a series of proton transfers (here drawn as the short, cheeky internal proton transfer) that creates an ammonium ion as a leaving group. Elimination of the amine forms an oxonium ion that is deprotonated to give the product.
Enamines are sufficiently nucleophilic that they will add to aldehydes to give, after hydrolysis, the product of the aldol reaction. The diagram below shows the synthesis of an enamine from a ketone and then reaction with a non-enolizable aldehyde. The first stage of the reaction, formation of the enamine, occurs with the release of water. This is followed by nucleophilic addition and formation of an iminium cation. Hydrolysis of the iminium ion requires water and gives the product. The mechanism of each step should be clear by now (but revision of the aldol reaction can be found HERE).

An amine-promoted aldol reaction. The reaction proceeds through the formation of an enamine that participates in nucleophilic attack on an aldehyde to give an iminium ion. Hydrolysis releases both the amine and the product. The interesting feature is that the amine is consumed while water is released at the start and then the amine is released and water consumed at the end. This suggests a catalytic cycle should be possible.
If you look closely at the reaction above you will notice that the amine is consumed at the start of the reaction and released at the end. At the same time, water is released at the start of the reaction and consumed at the end. This is balanced. This suggests that the reaction could be redrawn as a catalytic cycle … and I have:

The amine-mediated aldol reaction can be catalytic in both amine and a weak aqueous acid. The cycle in this reaction shows the consumption of pyrrolidine to form an enamine and its release once the aldol reaction has been performed. Slightly harder to see is water and a hydronium ion keep shuttling backwards and forwards so that neither are consumed in the full reaction.
In this catalytic cycle, all the steps of the mechanism are identical to those shown earlier. The only difference is that the cycle focuses on the amine. It interacts with the ketone to form an enamine that is then involved in the aldol reaction. Once this nucleophilic addition has occurred, there is an iminium carbocation that can be hydrolysed by water and acid to regenerate the amine. The oxonium species formed on the elimination of the amine is deprotonated to regenerate the acid as well. Both amine and acid can be used in a sub-stoichiometric quantity.
This catalytic cycle was key to the early examples of organocatalysis and ultimately, led to a whole new sub-discipline that exploded in the early 2000s, giving two of its proponents, List and MacMillan the Nobel Prize in 2021. It became clear that the aldol reaction was only one of a multitude of reactions that could be promoted by chiral secondary amines, such as the simple amino acid proline. Chiral secondary amines allow the control of stereoselectivity as shown in the example below:

Proline catalyzes the reaction of acetone, the nucleolphile, with an aldehyde as electrophile. The reaction only requires 30 mol% of the secondary amine (drawn badly in its non-zwitterionic form) to give a β-hydroxy ketone in good yield and high enantioselectivity.
While the origin of the stereochemistry is beyond this simple summary (one model involves hydrogen bonding and drawing a Zimmerman-Traxler chair-like six-membered transition state … there is a reason organic chemistry profs love those cyclohexane rings), you should be aware that the mechanism is exactly the same as the reactions above (*I know, there are still discussions in the literature as to the actual mechanism of these reactions, but from an undergraduate point of view, this is a good starting point).

Catalytic cycle for proline-mediated aldol reaction. A sub-stoichiometric quantity of amine is required. Water is expelled during the condensation step and consumed during the hydrolysis.
The diagram above shows a simplified version of the enamine-mediated aldol reaction that highlights both the regeneration of the amine catalyst and makes the role of water clearer. Water is formed during the condensation step that initially forms an iminium ion and is consumed in the hydrolysis of the second iminium ion, which regenerates the amine and gives the final product.
The chemoselectivity exhibited in this reaction is quite remarkable. Why doesn’t the nucleophilic amine react with the more electrophilic aldehyde first rather than making the enamine from the less reactive ketone? The simple argument involves steric hindrance/congestion. When the aldehyde and amine condense to form an iminium ion there is always a disfavorable interaction between the isopropyl group of the aldehyde and either a CH2 position or the carboxylic acid of proline. Reaction with acetone, the ketone is not hampered by this interaction.

The mixture of a ketone, an aldehyde and proline can lead to four different enamines. Two are formed from the aldehyde and are disfavored due to steric interactions between the methyl group of the double bond and proline. The other two are formed from the ketone. The favored conformation has the rigid double bond further away from the bulky carboxylic acid. All of this is important for the chemo- and stereoselectivity.
All this suggests that simple undergraduate chemistry is powerful enough to win a Nobel Prize.
Conclusion
Enamines are relatively stable enol, or enolate, equivalents. They are formed by the condensation of a secondary amine with an aldehyde or a ketone. The reaction is often chemo-, regio- and stereoselective.
Enamines are more reactive than enols and silyl enol ethers due to nitrogen being less electronegative, which allows for greater delocalization of the lone pair into the double bond. They are less reactive than charged species, such as enolates. Enamines will react with haloalkanes in simple SN2 alkylations, with enones in Michael or conjugate additions, and with aldehydes in the aldol reaction.
The mechanism shows that the secondary amine can be used as a catalyst as it is regenerated when the product is released. Sub-stoichiometric quantities of chiral cyclic amines, such as proline, are used in enantioselective aldol reactions, amongst other useful reactions. This was one of the observations that kick started the organocatalyst revolution. One day I’ll do a summary on organocatalysis.