Alkenes as nucleophiles: Part 1
Introduction
Non-conjugated alkenes are electron rich possessing reactive π electrons that make the alkene a nucleophile. Alkenes react with a range of electrophiles in electrophilic addition reactions. Some of these reactions are not true additions as the electrophile possess a leaving group but in all cases all the atoms of the alkene are found in the product.
I have classed these reactions as examples of π-nucleophiles as the key step will involve π electrons attacking the electrophile. What happens next in the reaction depends on the structure of the electrophile.
Addition of H–X
Alkenes react with hydrobromic, H–Br, and hydrochloric acid, H–Cl, in an addition reaction to give an alkyl halide. This is known as hydrohalogenation:

General reaction for the addition of H–X to an alkene.
The addition is a two step mechanism, and is shown below for addition of hydrobromic acid. First, the nucleophilic alkene attacks H–X. This is accompanied by loss a leaving group. You can either think of H–X being highly polarised by the electronegative halide resulting in attack on the partially positive hydrogen atom or that the leaving group must be the more electronegative atom (more stable leaving group). Either way, the electrons flow from the alkene to the proton and end on the halide. This results in protonation of the alkene and formation of a carbocation along with the bromide anion. This is an example of proton transfer.

The mechanism for the addition of HX to an alkene is a two step process involving proton transfer and then nucleophilic addition.
The second step is nucleophilic addition with the anion attacking the electron deficient cation to give the overall product of electrophilic addition. The carbocation is an sp2 hybridised carbon atom. This means it has trigonal planar geometry or is flat. The bromide anion nucleophile can add to either side of the flat carbon atom so there is no control over stereochemistry (or at least at undergraduate level).
When the alkene is non-symmetric, the halide can add to either end of the double bond giving two different products. One product is normally favoured and the reaction is said to occur with regioselectivity.
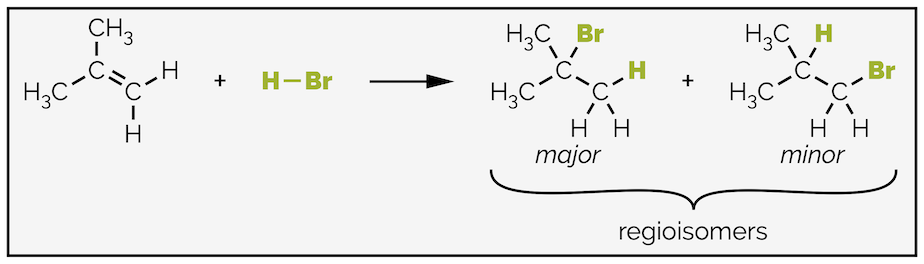
Non-symmetrical alkenes can lead to a mixture of regioisomers but invariably one isomer is favored.
The first step, protonation (proton transfer) of the alkene, determines the product. Addition of the hydrogen can give two different carbocations. Formation of the more stable carbocation is favored. The carbocation is stabilised either by an inductive effect, also known as hyperconjugation, or by resonance.

The regioselectivity of the addition of H–X to an alkene can be understood by the stability of the intermediate carbocation formed during the initial protonation step.
If you ignore resonance (which would be a foolish thing to do), then the stability of carbocations is that tertiary carbocations are more stable than secondary cations, which are more stable than primary cations, and methyl cations are the least stable (and not relevant to the addition reactions of alkenes). Alkyl groups are electron donating and the more electron donating groups that are attached to an electron deficient carbocation the more stable it is.

The stability of simple carbocations is determined by hyperconjugation or the inductive effect (two names for the same concept).
An aside (not needed at 1st year)
If you that want to understand why alkyl groups are electron donating you need to think of the orbitals present (for most first year courses, this is more information than you require). A carbocation is a carbon with only six valence electrons. It has an empty 2p orbital and a formal positive charge. Hyperconjugation describes the sharing of electrons from adjacent C-H or C-C σ bonding orbitals into this empty 2p orbital. It can be thought of as σ bond delocalisation. Effective sharing can only occur if the orbitals are parallel allowing full overlap. Only one bonding orbital per alkyl group can align. Thus more alkyl groups allow more hyperconjugation and hence greater stability.

Hyperconjugation is the delocalization of σ electrons by the overlap of σ bonds.
End of aside
The major product is often known as the product of Markovnikov addition. Markovnikov rule states that ‘the hydrogen adds to the carbon of the alkene that started with the most hydrogen atoms’ or simplified to ‘the rich get richer.’ The other regioisomer is known as the product of anti-Markovnikov addition or the anti-Markovnikov product. These terms are frequently used to describe the products of addition but the so-called Markovnikov rule is less useful as it doesn’t account for resonance. Instead of remembering a ‘rule’ it is better to understand the mechanistic reasoning and that the regiochemistry of addition is controlled by the preferential formation of the more stable carbocation intermediate.
Markovnikov’s rule cannot explain the following reaction as the ends of the alkene are indistinguishable if you are simply counting the number of hydrogen atoms:

Markovnikov's rule cannot predict the major product of this addition reaction but you can if you understand the mechanism of the reaction.
To understand the regioselectivity of this reaction, work through the mechanism. The first step is proton transfer and this can form two possible carbocations. Both are secondary carbocations but they are very different. One is resonance stabilized as the charge can delocalize over the aromatic ring. This is cation is more stable, and the reaction proceeds via this intermediate. More of the resonance stabilized cation is formed, the activation barrier for this pathway is lower. The second step, nucleophilic addition of the bromide anion gives the product.

Delocalization of the carbocation favors formation of the the top alkyl bromide.
One of the curly arrows used in the mechanisms shown above can cause confusion. The first arrow communicates that the π electrons are attacking the proton but it does not tell you which carbon will retain the electrons and form the new bond to hydrogen. There are a number of ways of addressing this, none of which have gained widespread acceptance (in other words, each has their supporters and detractors). The first, and probably clearest, is to show the new bond before it is formed as a dotted line (a partial bond; as shown in the first mechanism in this summary). The second is the atom specific curly arrow, here the arrow passes through the carbon atom that retains a share of the electrons. The final is the bouncing curly arrow, which bumps into the atom that forms the new bond. All are shown in the scheme below. I favor the atom specific curly arrow but tend to draw the partial bond in these summaries as it doesn’t require this explanation!

Different ways of representing the redistribution of electrons during the proton transfer step so that it is clear which carbon atom retains a share of the electrons.
Taking things a little further …
The details above are sufficient for most first year (100 level) chemistry courses, but some readers might be interested in the orbital interactions that lead to the addition of H–X across an alkene. These are shown in the scheme below. The proton transfer occurs when highest occupied molecular orbital (HOMO) of the nucleophile, the π bond of the alkene, overlaps with the lowest unoccupied molecular orbital (LUMO) of the electrophile, the σ* antibonding orbital of H–X. The electrons move from a π bond to a new C–X σ bond. By filling the σ* antibonding orbital the H–X bond is broken. The second step involves the overlap of the HOMO of the bromide anion, a non-bonding orbital or lone pair, with the LUMO of the cation, the empty 2p atomic orbital. The overlap can occur from either side and this leads to a loss of stereochemical information.

The frontier orbitals involved in the addition of H–X across an alkene. The two steps involve different orbital interactions.
While an understanding of the orbital interactions is not necessary for students not taking chemistry further, I hope that it is clear that it can provide a rationale for the observations that aren’t fully explained by using curly arrows (or horror of horrors, avoids having to learn facts by rote).
End of extra info.
Hydration of Alkene, the Addition of H2O
Acid catalyzed addition of water across an alkene, otherwise known as the hydration of an alkene, leads to the Markovnikov addition product. The reaction is almost identical to the hydrohalogenation above just with X equivalent to OH instead of a halide.

The acid catalyzed hydrogenation of an alkene. The reaction is similar to hydrohalogenation and shows the same regioselectivity.
The mechanism starts with proton transfer or protonation of the alkene. The proton source will be protonated water, H3O+, the hydronium species. As before, proton transfer gives the most stable carbocation and this controls the regioselectivity. It will lead to Markovnikov addition.
The second step is nucleophilic addition of water to the highly electrophilic carbocation to give a new oxonium species. There is a second proton transfer that gives the product alcohol and regenerates the hydronium catalyst.
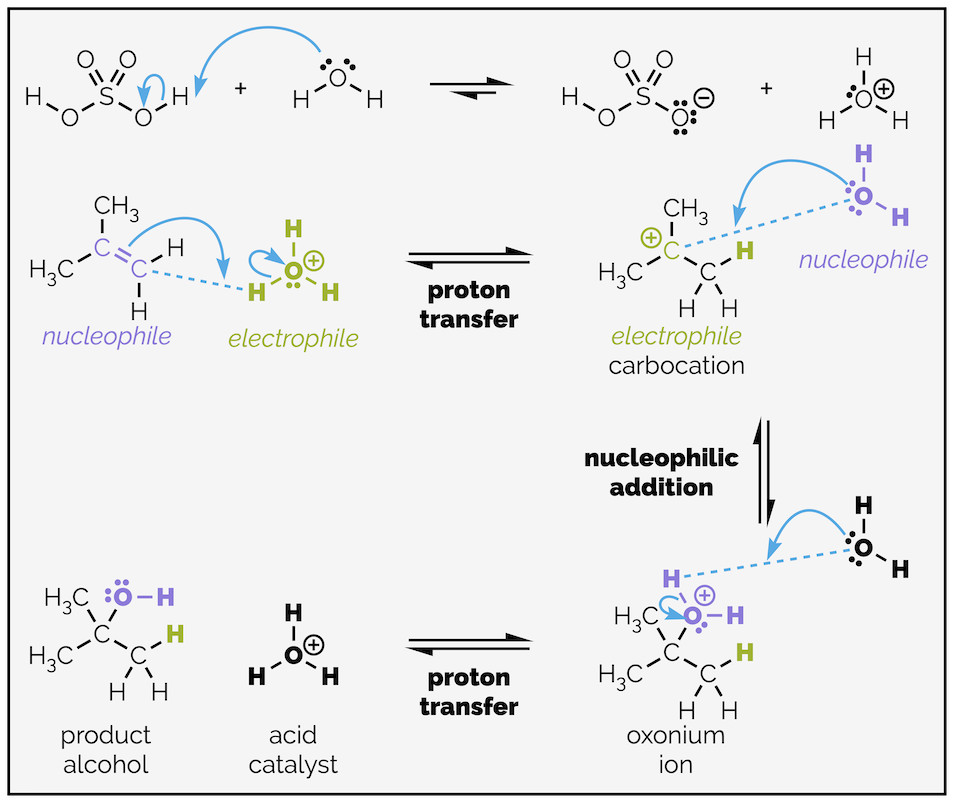
Mechanism for the acid catalyzed hydration of an alkene. The acid adds one step (the second proton transfer), otherwise it is identical to hydrohalogenation.
As with hydrohalogenation the reaction is regioselective but not stereoselective. The formation of the trigonal planar carbocation means the nucleophilic water is capable of attacking either side equally well (alright, this isn’t always true but substrate control is a topic for a more advanced summary, and many students will never need to know!).
What is useful to know is that the reaction arrows are equilibrium arrows, indicating that each step is reversible. This is not a mistake. It is possible to perform an elimination reaction, converting an alcohol into an alkene. To force the reaction in the desired direction it is necessary to disrupt the equilibrium (Le Chatelier's Principle). Using dilute acid leads to the hydration of an alkene. Dilute acid has an excess of water present and this will act as a nucleophile (effectively you are increasing the amount of starting material so the reaction counters this by forming more product). If you take an alcohol and react it with concentrated sulfuric acid then elimination occurs. Concentrated acid has little or no water present and, in the case of sulfuric acid, is a good drying reagent, readily forming hydrogen bonds to water. Removing water from the left-hand side of the reaction results in the reaction shifting to the left. The mechanism is the reverse, starting with protonation of the alcohol then loss of a leaving group. The formation of alkenes will be in a different summary.
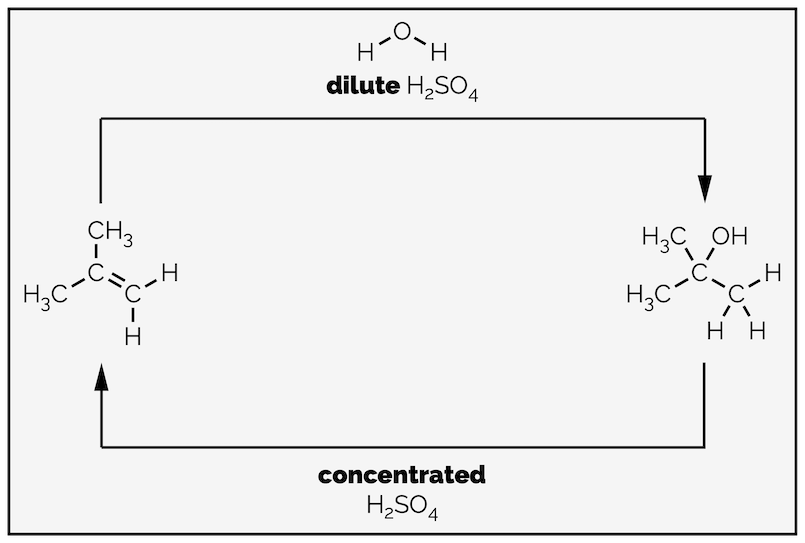
Hydration of an alkene is a reversible reaction. Using dilute acid, so that there is an excess of water, leads to hydration. Using concentrated acid, with no water present (concentrated sulfuric acid is a drying reagent), leads to elimination and conversion of an alcohol into an alkene.
People don’t like treating their precious compounds with acid (or forming carbocations as these often rearrange … which would make a good topic for another summary) so other methods for the hydration of alkenes have been developed. I’m just going to comment on one that leads to Markovnikov addition (anti-Markovnikov addition and hydroboration/oxidation is the subject of one of the next few summaries) and is frequently found in organic chemistry textbooks, oxymercuration-reduction. I don’t know if this chemistry is actually used much these days as mercury salts can be unpleasant (although I did do this reaction during my PhD and remember enjoying watching mercury metal drop out of the reaction).
In this reaction, the alkene is treated with mercury acetate, Hg(OAc)2, and water followed by sodium borohydride, NaBH4, and it gives the same alcohol as before (the Markovnikov product).

Oxymercuration-reduction (or oxymercuration-demercuration) results in the Markovnikov addition of water across an alkene. The reaction is milder than acid-catalyzed hydration and does not involve the formation of a carbocation that could undergo rearrangement.
The mechanism is different but still involves the alkene acting as a nucleophile. The first step is formation of a mercuronium species, a positively charged three-membered ring containing mercury. The triangle is deliberately drawn distorted so that there is a longer bond on one side. The commonly drawn curly arrows show the alkene attacking the Hg2+ salt and kicking out an acetate as a good leaving group. A lone pair of electrons on the massive mercury ion attacks the alkene before a carbocation is formed.
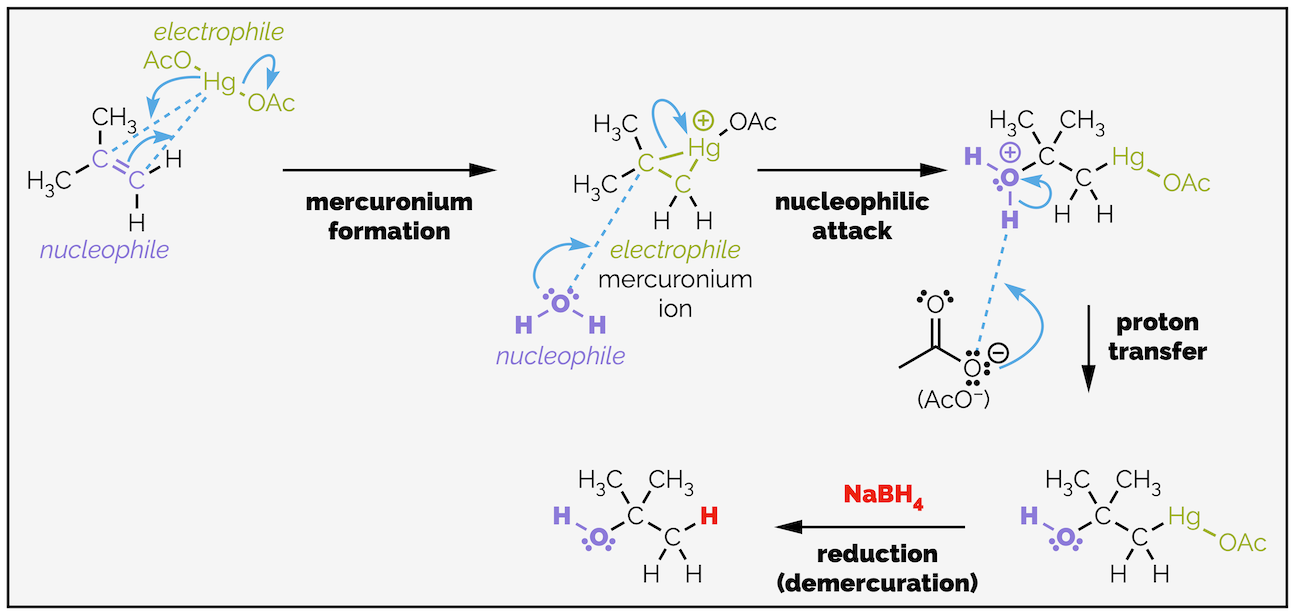
Oxymercuration involves formation of a charged mercuronium ion followed by attack of a nucleophile to give an organomercury species. Reduction leads to demercuration and the formation of the Markovnikov product.
The cationic mercuronium species is highly reactive, and water, or another nucleophile, will attack one of the carbon atoms causing a C–Hg bond to break. Effectively, this is loss of a leaving group. The nucleophile attacks from the opposite face to the huge mercury species. This is a result of orbitals (and I’ll cover substitution reactions in a different summary) but can also be visualized as the nucleophile avoiding the steric bulk of the mercury. In theory, this means the reaction is stereospecific but this is normally negated in the reduction step. The attack is regioselective and occurs at the more substituted end of the mercuronium species. This end can stabilize a carbocation more readily, or at least the build up of positive charge and I’ll discuss this in detail below in the section on bromohydrin formation (see below).
At the end of the hydration reaction, a reducing reagent, normally sodium borohydride, is added to the reaction and this removes the mercury. The reaction involves formation of a mercury hydride and then elimination. The reduction occurs with loss of stereochemical information (the hydrogen can add to either side of the carbon atom).
There are strong similarities between oxymercuration and the formation of bromohydrins (or iodohydrins). As the latter are far more common, I’ll discuss them in more detail below.
Bromination
A classic test for the presence of an alkene used to be the addition of bromine, Br2. A red solution of bromine would be added to a colorless solution of the compound to be tested, if the color vanished, then an alkene was probably present. If the color persisted then it was unlikely that an alkene was present. The test worked as solutions of bromine are highly colored while most alkyl bromides are not. An example is given below:

The bromination of alkenes was a standard test for alkenes. A red-brown solution of bromine (either neat or in solution) would go colorless when added to a solution containing an alkene. The reaction is an addition reaction with bromine adding across the alkene.
The reaction is an addition reaction with all the atoms of the starting materials found in the product. There is no issue of regioselectivity as bromine atoms add to each end of the alkene. Stereospecificity is important. The two bromine atoms always add to opposite sides of the alkene. This is known as anti-addition. If the bromine atoms were on the same side it would be an example of syn addition.
It is easy to visualize anti addition with cyclic molecules, such as cyclohexene in the example above, as one bromine is on the top face (a wedged bond) and the other on the bottom face (a hashed bond). It is also possible to observe anti addition with acyclic alkenes and this becomes more pronounced if you compare the bromination of different stereoisomers (diastereomers or geometric isomers).
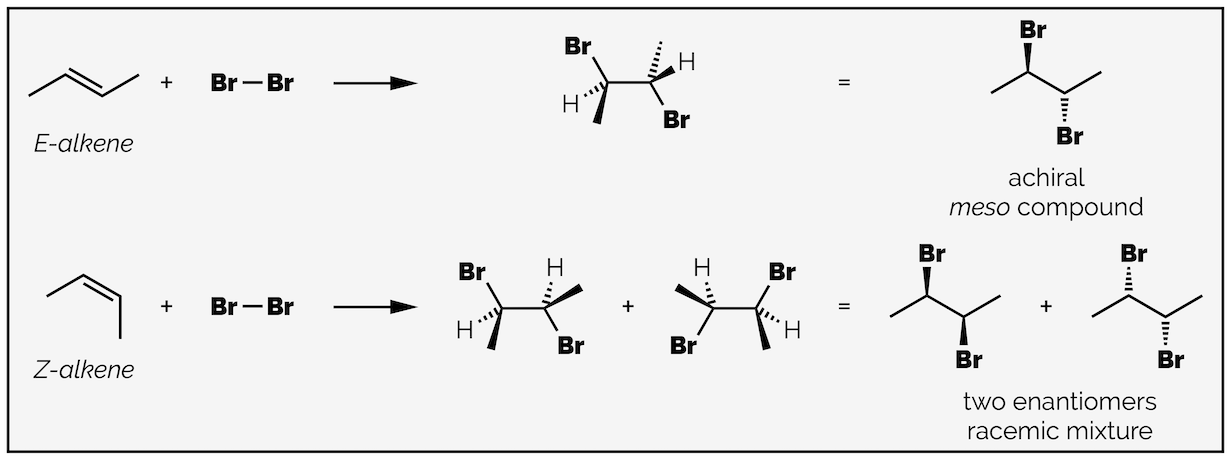
The anti-addition of bromine to the two diastereoisomers of an alkene will give different stereoisomers. Addition to the E-alkene leads to a single compound, the achiral dibromide while addition to the Z-alkene gives two enantiomers of the dibromide as a racemic mixture.
Both reactions above proceed with anti addition. In both cases, the Br atoms add to opposite faces of the alkene. I have tried to make this clearer by showing the molecule in a conformation that has the two bromine atoms in the same plane but on opposite sides of the central C–C bond. In the top reaction, addition to the E-alkene, a single product is formed. This is an achiral compound. It might not be instantly clear why it is not chiral but if you rotate the central C–C bond to give a second conformation you will see that there is an internal plane of symmetry. This means the molecule is identical to its mirror image. Achiral compounds with multiple stereocenters and an internal plane of symmetry are called meso compounds.

Changing the conformation of the dibromide reveals that it has an internal plane of symmetry. Molecules with a plane of symmetry cannot be chiral. As the molecule has two stereocenters yet is achiral it is known as meso.
Anti addition to the Z-alkene gives two non-superposable mirror image dibromides. These are enantiomers. There is no selectivity in the reaction so a 50:50 mixture or racemic mixture of the two is formed.
Before discussing the mechanism, I want to highlight some terminology. In chemistry, there is a difference between a reaction being selective or being specific. A stereospecific reaction is different to a stereoselective reaction. And, because chemistry hates you, a stereospecific reaction can be stereospecific but it doesn't have to be. The above example is stereospecific but not stereoselective. Other brominations might be both! The mechanism of a stereospecific reaction means that a specific stereoisomer must be formed. In this case, it means the anti diastereomer is always formed. Other stereospecific reactions include SN2 substitution, where there must be inversion of stereochemistry. A stereoselective reaction can give a mixture of stereoisomers but one might be favored. Nucleophilic addition to an aldehyde with an α-stereocenter can be stereoselective but is not stereospecific as the nucleophile can add to either face of the carbonyl group. It may be 100% selective but it is never stereospecific. The bromination above is stereospecific as it must give the anti dibromide but it is also non-stereoselective as both enantiomers are formed. Sometimes I just love terminology.
The stereochemical outcome of the bromination of both the cyclic alkene and the acyclic geometric isomers tells you that there must be anti addition. The mechanism of the reaction must explain this, and so this results gives a big clue as to what is happening during addition (and the fact the addition must occur in two steps).
The first step of the mechanism is nucleophilic attack of the alkene on the bromine to form a bromonium ion. This is shown by two electrons of the nucleophilic π bond attacking the bromine. The attack is accompanied by the loss of a leaving group, an electronegative bromine departs as a bromide anion. At the same time, a lone pair of electrons on the large bromine atom attack the other end of the alkene to form a three-membered ring. The result is a highly electrophilic, positively charged species known as a bromonium ion. This initial step is concerted. This means both C–Br bonds are formed at same time as the C=C bond breaks. The reaction is reversible. The bromide can attack the bromine atom of the bromonium species and reform the starting materials but as the second step is irreversible this is frequently ignored. Unlike the hydrohalogenation above, a carbocation is never formed (this would be a stepwise process). The fact that anti-addition is always observed and that the two geometries of the acyclic alkene give different diastereoisomers tells us that the carbocation is never formed.

The first step of the addition of bromine to an alkene is formation of a bromonium ion.
Bromine is a non-polar molecule as both ends of the bond are identical. So, why does it behave as an electrophile? Bromine atoms are big, and their outer electrons are far from the nucleus. This makes them easy to polarize, or they are said to be highly polarizable or have a high polarizability. As the bromine approaches the electron rich alkene, the π electrons repel the electrons of bromine. This makes one end partially positively charged. It is now electrophilic and the nucleophilic alkene can attack. (As always, this explanation appears to be a simplification. It is sufficient for most undergraduate courses but further down this summary, I'll show you that kinetic experiments suggest that a polarized Br2 molecule is not the actual electrophile).
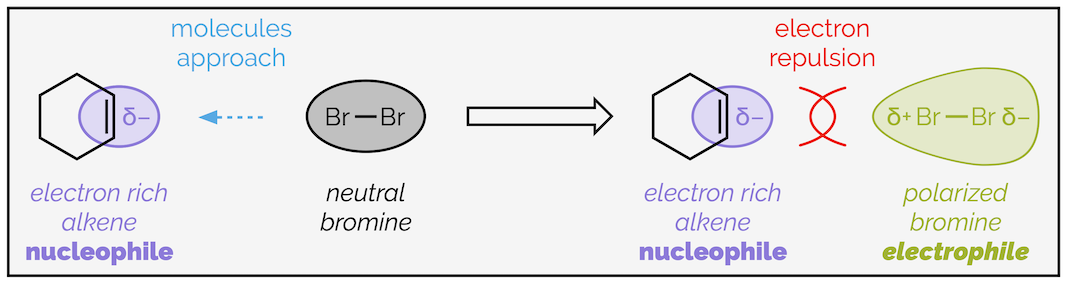
The polarization of bromine by interaction with an electron rich alkene.
The standard curly arrows for this break our normal guidelines for curly arrows and have two arrows moving in opposite directions. This isn't ideal but gives, in my opinion, a clearer idea of the bonds being formed and broken. Curly arrows are simply a description of the rearrangement of electrons during a reaction, they are not what is happening. If you are interested in a better understanding of the interactions between alkene and bromine, read the section below discussing the orbital interactions. For most undergraduate courses, you don't need to go into that much detail.
The second step of the addition involves the nucleophilic attack of the electron rich bromide anion on the electrophilic bromonium cation. Addition occurs from the opposite face to the bulky bromine atom to minimize interactions between the large bromine atoms. This results in anti addition. There is also a stereoelectronic explanation for the anti attack, and I'll discuss the orbitals later (this is effectively an SN2 reaction and must occur with backside attack for those of you that have already studied substitution reactions).

The second step of the addition reaction is nucleophilic attack of the bromide on the bromonium ion. This occurs from the opposite face to the first bromine atom and leads to the anti-dibromide. This is shown in three different representations above.
The two enantiomers are formed as the bromide anion can attack either end of the bromonium ion. You could also argue that the initial bromonium species could form on either face of the alkene but this actually gives the same molecule drawn in two different ways as there is a plane of symmetry (as long as you ignore the conformation of the molecule). Again, a meso compound has been formed.
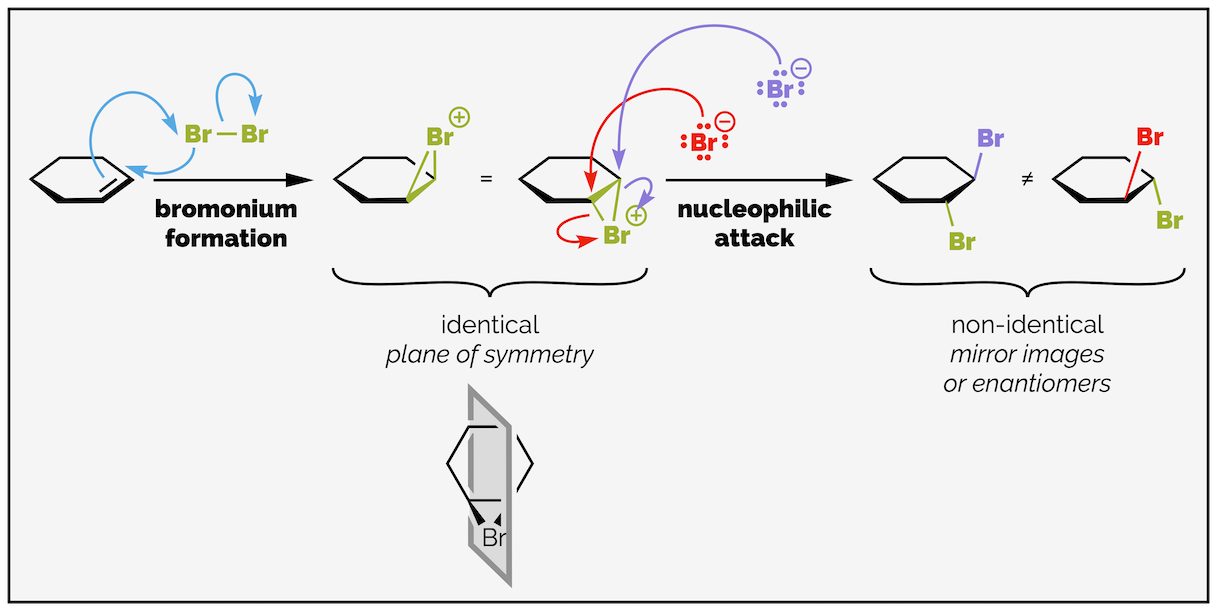
The formation of enantiomers is the result of the bromide anion attacking either end of the alkene.
The overall mechanism for the addition of bromine to an alkene is given below. There are two discreet steps; formation of the bromonium ion and then nucleophilic addition leading to anti addition. We know the bromonium ion forms as it can be isolated if extreme care and very sterically demanding alkenes are used. Formation of the bromonium ion is reversible but addition of the second bromine atom is irreversible, driving the reaction to completion. Reversibility of bromonium formation (and iodonium formation) is very useful in stereoselective cyclizations but that is a story for another day (probably postgraduate study).

The two step mechanism for the addition of bromine to an alkene.
I know I’m repeating myself, but this point is important in terms of the mechanism and the stereochemical outcome of the reaction but the observation that bromination gives the anti-product indicates that a stepwise mechanism with a carbocation intermediate (shown below in lurid red) is not occurring. If the reaction proceeded through a carbocation intermediate then the nucleophilic bromide anion could attack from either face to give both the syn and anti product or the central C–C bond could rotate, again giving a mixture of diastereoisomers. So this is not the mechanism:

The bromination of an alkenes does not proceed via the formation of a carbocation.
Taking things a little further …
As with the hydrohalgenation and hydration of alkenes, the curly arrow mechanism is enough for most undergraduate courses, and you can skip ahead to bromohydrin formation. A better understanding of the mechanism can be obtained by thinking about the orbitals involved in the reaction. The first step, formation of the bromonium ion, involves two sets of interactions as two new C–Br σ bonds are formed. Effectively, one set of interactions describes the alkene acting as a nucleophile while the other set of interactions treats the alkene as the electrophile.

The orbital representation of bromonium ion formation. The HOMO of the alkene attacks the bromine while the LUMO of the alkene is attacked by the bromine.
The HOMO is the alkene π bond, and this overlaps with the LUMO of the bromine molecule, which is the Br–Br σ* antibonding orbital. This interaction leads to the formation of a C–Br bond and the breaking of the Br–Br bond. It is also why some textbooks do not include the third curly arrow showing the formation of the second C–Br bond. This second is made by the overlap of a non-bonding lone pair of electrons on the bromine with the π* antibonding orbital. This gives the bromonium species and leads to the π bond breaking. The interactions are more or less simultaneous, occurring when the two molecules are sufficiently close. This accounts for the concerted nature of the reaction.
The second step of the reaction is the attack of the bromide anion on the the bromonium ion. The bromide anion is the nucleophile with the HOMO being a non-bonding lone pair of electrons. The LUMO of the bromonium species is a C–Br σ* antibonding orbital. This has a larger coefficient at 180° to the bond and this explains why there bromide must attack from the opposite face and we observed anti-addition.

The orbital interactions for the second step of bromination, nucleophilic attack by the bromide on the bromonium species, explain why anti-addition is observed. The nucleophile must approach at 180° to the C–Br bond.
Another factor often missed from textbooks is the fact that the experimental kinetics of the bromination don't match the mechanism given in textbooks (which is naughty, as much of our mechanistic understanding comes from kinetics experiments). It appears that bromination is second order with respect to bromine. That means there are two molecules of bromine involved in the reaction and it is thought these interact first to create the real electrophile. This is shown in the scheme below:

Kinetic experiments suggest that the brominating reagent is bromonium ion formed from the reaction of two equivalents of bromine.
End of the extra info.
Bromohydrin Formation
The bromonium ion formed during the addition of bromine is highly electrophilic and other nucleophiles can add instead of the bromide anion. One example of this is the use of bromine in water (or an alcohol). Under these conditions, a molecule containing both a bromine atom and a hydroxyl group is formed. This is called a bromohydrin.

A mixture of bromine and water will add to an alkene to give a bromohydrin. The reaction is stereospecific for the anti-diastereoisomer and regioselective for addition of the alcohol to the most substituted end of the alkene.
The example above uses a cyclic alkene to show that the reaction is stereospecific, giving the product of anti-addition, with the bromine atom and the alcohol on opposite sides. This occurs as the mechanism is the same bromination except that it water acts as the nucleophile in the second step.

The mechanism for bromohydrin formation is the same as bromination except that there is an additional proton transfer step.
Step 1 is formation of the bromonium ion with the nucleophilic alkene attacking the bromine electrophile. Formation of the bromonium ion and not a carbocation means that water must add to the opposite face in step 2. This is nucleophilic addition. This forms the oxonium species that is deprotonated in step 3.
Why does water attack and not the bromide ion? This is a good question as the bromide ion is the better nucleophile (it is both more electron rich and more polarizable than an oxygen atom, factors that make it a better nucleophile). The answer is that the bromonium ion is highly unstable and will react with the first nucleophile that it can. If you perform the reaction in water there is more water present and the chance of it attacking instead of Br– is greater..
The mechanism above, and the explanations of selectivity below are all applicable to oxymercuration. The reactions are effectively the same, the only difference is that there is a demercuration step. Arguments of regio- and stereo-selectivity are the same.
Why does the nucleophile attack the more hindered carbon of the bromonium species in step 2 of the mechanism? Why is the reaction regioselective (the nucleophile preferentially adds to one end even though the mechanism would allow it to add to either)? There are two useful explanations to this observation. The first suggests that the C–Br is starting to break before the nucleophile attacks. If you like, the bromonium ion is not a perfect equilateral triangle as drawn above but actually has one bond longer than the other. This leads to a build up of partial positive charge on a carbon atom. In non-symmetric bromonium ions, the carbon that stabilizes the partial positive charge better is favored. This is the same carbon that could stabilize a carbocation or the more substituted carbon. The nucleophile attacks this more electrophilic carbon.

The regioselectivity can be explained by considering that the C–Br starts to break before the nucleophile attacks. This leads to the build up of a partial positive charge on a carbon atom. The carbon atom that best stabilizes a carbocation, in this case the tertiary carbon, best stabilizes the partial positive charge.
This argument can almost be viewed as through resonance. You could imagine a series of resonance structures that involve the breaking of a σ bond as shown below. This means the positive charge can be on the bromine or either carbon atom. The resonance structures that stabilize the carbocation best contribute the most to the resonance hybrid, the real structure. Again, this means we can ignore the structures that have the carbocation on a primary carbon. I don't like this argument as students start to think the carbocation actually forms, it does not.

It is possible (but I don't like it) to argue the selectivity with 'resonance' structures.
The second argument to explain the regioselectivity and this considers the stability of the transition state. When the nucleophile attacks the bromonium ion, the positive charge is effectively transferred from the bromine atom to the nucleophilic oxygen atom. The transition state describes the highest energy arrangement of atoms that includes both starting materials and products. The new and old bonds are being partially made and partially broken. This means the charge is spread out over three atoms, Br–C–O. The two possible transition states are shown below. The more stable transition state (lower in energy) has the more stable partial positive charge. The reaction will proceed by the lower in energy pathway or this transition state.

The regioselectivity can be explained by considering the transition state of the reaction, this has a charge being redistributed across two atoms. One transition state allows this transfer to occur via the more stable tertiary position while the other requires the build up of positive charge on a disfavored primary position.
Another form of selectivity has to be considered and that is chemoselectivity (or possibly site selectivity). This describes how different functional groups, or in this case, different alkenes react at different rates. If there is a sufficient difference in the rate of reaction then one group will react before the other and you have another example of selectivity. In all these reactions, the alkene is acting as a nucleophile. The more electron rich an alkene, the more nucleophilic it is, and the faster it will react. This means the more substituted an alkene (as long as the substituents are simple alkyl groups) the more reactive it is. Each substituent is electron donating through hyperconjugation so the more groups the more electron rich the alkene.

When the rate of reaction of four different alkenes is compared, you can see that the more substituted the alkene the more reactive is due to increased nucleophilicity. The tetra-substituted alkene is almost 2 million times more reactive than the unsubstituted alkene.
This last example reveals that other nucleophiles will react with the bromonium ion, not just bromide anions or water. Any good nucleophile will react if it is in high enough concentration. The nucleophile can be part of the original molecule, and it is possible to active an alkene as a bromonium ion (or more commonly, activated as an iodonium species) so that the molecule can cyclize.

An example of bromolactonization. This is exactly the same as the bromohydrin reaction above. The alkene is activated by formation of a bromonium ion then the nucleophilic carboxylate anion participates in nucleophilic attack, opening the bromonium species and giving the cyclic ester or lactone.
Epoxidation with Peracids
A related reaction is epoxidation using peracids (also called peroxy acids). This involves the addition of a single oxygen atom to an alkene. I’ve included it in this summary as the alkene is acting as a nucleophile, and because the mechanism shares important similarities with bromination. Peracids are carboxylic acids with an additional oxygen atom between the carbon of the carbonyl and the OH group. They are only weakly acidic as there is no delocalization of the negative charge (O–) upon deprotonation. They are good electrophiles due to a weak O–O bond and one end being a good leaving group. The most common peracid is meta-chloroperoxybenzoic acid or m-CPBA. It is reactive, the electron withdrawing aromatic ring enhancing the electrophilicity but, most importantly, it isn’t explosive (as long as it is kept wet or in solution). Epoxidation with a peracid is shown below:

An example of epoxidation with a peracid, in this case the common reagent meta-chloroperoxybenzoic acid.
Epoxidation is similar to the formation of the bromonium ion as it is a concerted reaction, with all bonds being made and broken at the same time (more or less). The concerted nature means the reaction is stereospecific, with both new C–O bonds being formed on the same face of the alkene. This is known as syn-addition. This can be seen in the cyclic example above, and is more evident when you look at the epoxidation of the two geometries of an acyclic alkene:
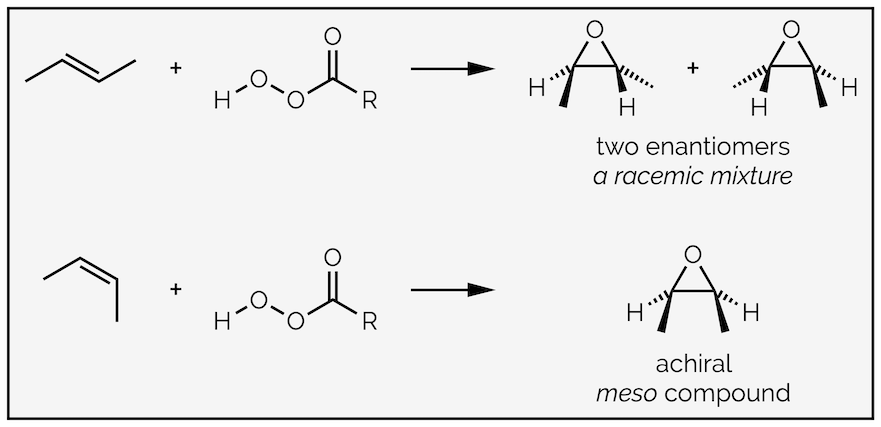
The peracid-mediated epoxidation of acyclic alkenes occurs with retention of stereochemistry as the reaction is concerted.
The cis and trans alkenes give different stereoisomers. Epoxidation occurs with retention of stereochemistry as the trans alkene gives a racemic mixture of trans epoxides, while the cis alkene gives the meso cis epoxide. The internal plane of symmetry that makes the cis epoxide achiral should be easy to visualize.
The retention of stereochemistry tells you that the reaction is concerted and that the two C–O bonds are made at the same time. If the reaction had been stepwise with a carbocation intermediate then there would be a loss of stereochemical information. Any mechanism must take this into account.
The alkene is the nucleophile and the π electrons attack the electronegative oxygen atom. This oxygen is more reactive as it is attached to an electron withdrawing oxygen and carbonyl group. Breaking the O–O bonds allows the a good leaving group, the resonance stabilized carboxylate anion, to be kicked out.

Peracid-mediated epoxidation is a one step, concerted reaction, with all the bonds made and broken at the same time.
To form the second C–O bond, you need to follow the flow of electrons. As the O–O is broken, the electrons move to make a new carbonyl bond. The original carbonyl now breaks and takes the proton of the peracid to form a carboxylic acid. The electrons of the O–H bond are redistributed to make the C–O bond and give the epoxide. As all bonds are made and broken at the same time, the two C–O bonds must on the same face. The curly arrows look like a mess but as long as you draw the reagents with the correct orientation, the curly arrows flow round. This is the accepted mechanism found in most textbooks. Those who want more detail should consider the orbitals (I’ll discuss these below) and at that point it becomes clear that the mechanism is a little subtler than stated (in other words, it isn’t correct).
By studying the rate of epoxidation it is clear that the alkene is reacting as a nucleophile. The more substituted the alkene the faster the reaction. The reason for this is that each substituent is electron donating, and so the more substituents the more electron rich the alkene and the more nucleophilic it is.

The more substituted the alkene, the more electron rich it is, and the more nucleophilic. The more nucleophilic the alkene the faster the epoxidation.
Taking things a little further ...
As with the other sections, I have already covered all the information most students taking an organic chemistry course require (unless you are doing a chemistry degree). Read on if you are interested in the orbitals involved.
Like formation of the bromonium ion, there are two sets of orbital interactions. This is necessary as two bonds are being formed. The first involves the HOMO of the alkene, this is the π bonding orbital. This will overlap with the LUMO of the peracid. This is the O–O σ* antibonding orbital. Sharing electrons between a carbon atom and the oxygen will lead to the formation of a new C–O σ bond. At the same time, the electrons entering the antibonding orbital will break the O–O bond.

The orbital interactions for the formation of an epoxide with a peracid are almost identical to those found in the formation of the bromonium ion. The same concepts and patterns are used repeatedly in chemistry and there isn’t half as much to remember/understand as many students think.
At the same time, there is a second interaction. This is between a lone pair of electrons on the oxygen that are a sp3 hybridized non-bonding orbital on the oxygen atom and the π* antibonding orbital on the alkene. This overlap results in the formation of a second C–O σ bond and breaking the π bond of the alkene.
It is the lone pair of electrons that attacks the alkene. Non-bonding electrons are normally in higher energy orbitals than bonding electrons (this is why the bond formed in the first place, to make something more stable). The oxygen is acting as the nucleophile in this interaction and the antibonding orbital of the alkene is the electrophile. This means the curly arrow mechanism given above (and in every textbook I have read) is wrong, the electrons of the O–H bond are not forming the new bond. The mechanism almost certainly has two steps, with a hidden protonated intermediate. There is an informative discussion of this on Henry Rzepa's blog.
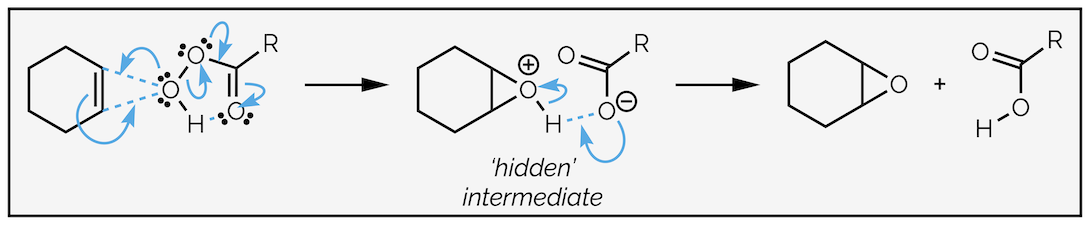
The real curly arrows for epoxidation should probably be drawn more like this. The key difference is that the oxygen lone pair of electrons is used to form the second C–O bond of the epoxide. The arrows also reveal another dirty secret of organic chemistry, the orbital alignment of the σ bonds means they can’t form the π bond so we need more arrows. The simplified version is much nicer and no one ever said curly arrows were real!
End of extra information
Syn-Dihydroxylation
The last reaction I want to cover is syn-dihydroxylation, the addition of two oxygen atoms to an alkene. This reaction is very different addition to bromination as you shall see when we look at the stereochemical outcome and then the mechanism (or a bit of it). The general reaction is:

Dihydroxylation with osmium tetroxide.
Syn-dihydroxylation is a stereospecific reaction that gives the syn-diol (surprise, it is in the name after all). A number of reagents can be used but the most common is osmium tetroxide. Osmium tetroxide is an unpleasant (and expensive) reagent and it is normally used as a catalyst in substoichiometric quantities. A second oxidant is added to regenerate the reactive species. This is often NMO or N-methylmorpholine-N-oxide in a reaction known as the Upjohn Process.
The syn-addition can be seen with oxidation of acyclic alkenes. The reaction must be concerted as the two alcohols are always on the same face (there is no cationic intermediate that allows the central C–C bond to rotate).

The dihydroxylation of an alkene with OsO4 is stereospecific but it gives the opposite results to bromination. Dihydroxylation gives a racemic mixture with the E-alkene while bromination gave the racemic mixture from the Z-alkene. Dihydroxylation is a syn addition while bromination is an anti addition.
The retention of stereochemistry reveals that the reaction is concerted with the two oxygen atoms being added across the alkene at the same time. That the dihydroxylation of the E-alkene gives a mixture of enantiomers while bromination of the same alkene gave the meso compound shows that this reaction occurs with syn addition and not anti addition. As always the mechanism must match this observation.
The mechanism involves a cycloaddition with the alkene attacking the tetrahedral osmium(VIII) reagent. The curly arrows show the osmium being reduced (the addition of electrons) to osmium(VI) and the second C–O bond being formed in a cyclic flow of electrons. This results in the formation of an osmate ester. It is an example of a true addition reaction. The reactions are performed in the presence of water and the osmate ester can be hydrolysed in an analogous fashion to an organic ester. This gives the diol and an osmium(VI) species. The latter can be re-oxidized by NMO to reform the reactive osmium(VIII) species, and in this way the reaction can be rendered catalytic with respect to osmium.
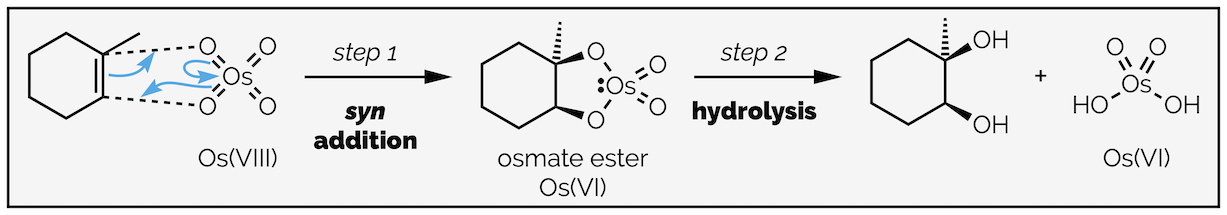
A simplified version of the mechanism for syn-dihydroxylation of alkenes.
Conclusion
Isolated alkenes are good nucleophiles. They are electron rich with four electrons shared between two carbon atoms. In all the addition reactions above the first curly arrow is the alkene attacking an electrophilic reagent. What subsequently happens depends on the electrophile and whether it contains a leaving group or its own nucleophilic component.

The first step of all these reactions involves the alkene behaving as a nucleophile and attacking another reagent.
A number of addition reactions have been described. Hydrohalogenation and hydration are the addition of H–X (X = Cl, Br, I or OH) across the alkene. These additions proceed by a stepwise mechanism and are regioselective but not stereoselective or stereospecific. The key to determining the regioselectvity is understanding what factors stabilize a carbocation (hyperconjugation and delocaliazation). This is sometimes known as Markovnikov addition.

A summary of the reactions covered in this summary (so a summary of a summary)
Bromination is also a stepwise reaction but now it is stereospecific and it always gives the anti product. As the same atom adds to both ends of the alkene, regioselectivity is not important. A modification of bromination allows the formation of bromohydrin or related compounds. The mechanism of the reaction is the same as bromination and is stepwise. It is stereospecific and regioselective. Epoxidation is a concerted reaction that occurs with syn-addition. It is stereospecific while regioselectivity is not an issue. Finally, syn-dihydroxylation is essentially a concerted process (ignoring the subsequent hydrolysis) that delivers syn-diols. It is stereospecific.
In the coming summaries I will discuss how we can reverse regioselectivity and achieve anti-Markovnikov addition. I will also discuss more stereoselectivity.