Anti-Markovnikov Addition
Introduction
Isolated alkenes are electron rich and are good nucleophiles. They undergo a number of addition reactions. Two important examples are the hydrohalogenation of alkenes and the acid catalyzed hydration of alkenes.

Hydrohalogenation and acid-catalyzed hydration of alkenes favors Markovnikov addition.
These reactions follow a similar mechanism and are regioselective. They favor the formation of the so-called the Markovnikov addition product or proceed with by Markovnikov addition. This means the new functionality, the halogen or alcohol, adds to the more substituted end of an alkene. The mechanism of explains this observation with proton transfer or protonation occurring to give the most stable carbocation.
How do you reverse the selectivity and add functionality to the opposite end of the double bond? How do you form the anti-Markovnikov product? This summary tells you (surprise).
This summary just covers the basic ‘textbook’ reactions for reversing Markovnikov selectivity. There are other elegant methods in the literature but I’m trying to cover the basic concepts here.
Anti-Markovnikov Addition of HBr
The standard Markovnikov addition product is obtained by an ionic reaction that is controlled by the polarization of bonds. By performing the reaction under radical conditions the regioselectivity is reversed. This is commonly achieved by the addition of a radical initiator.
Radicals, and radical reactions in organic chemistry are a fascinating subject. There will undoubtedly be a summary dedicated to them (I used to write an annual review on radicals so quite like them!). Below is a very simple introduction that is focused on the reversal of regioselectivity.

When the addition of H–Br is performed in the presence of a radical initiator, such as a peroxide, the anti-Markovnikov product is favored.
The addition of a peroxide, RO–OR, with a weak O–O bond, encourages the formation of radicals, species with a single unpaired electron. This reverses the normal selectivity of the reaction and favors the formation of the anti-Markovnikov product. This reaction works reliably for the addition of H–Br but not the other hydrohalogens. While I'm going to cover a new reaction mechanism, the principles that determine the regioselectivity of this reaction are the same as discussed in previous summaries.
The radical addition follows a radical chain reaction. It is a chain reaction as, in theory, you only need to create a single radical in order to transform all your starting materials to products as the key reactive species gets reformed continuously. The reaction commences with the formation of the first radicals, this is known as the initiation step. The example below involves dibenzoyl peroxide as the radical initiator, and leads to breaking the weak O–O bond. This is achieved by heating or using light (hν).

The first step of a radical chain reaction is initiation. This is formation of the initial radical and creation of the radical chain carrier, the species the will propagate (perform) the productive part of the reaction.
The relatively weak O–O bond undergoes homolytic scission (or fission, cleavage or homolysis), a fancy way of saying that a bond breaks and the electrons are equally divided between the two products. Breaking the bond in such a fashion gives two oxygen centered radicals or atoms with seven valence electrons. The chemistry of radical reactions is largely driven by the strength of bonds, each step occurs to give a stronger bond. So after the initial homolytic cleavage, each step breaks a weak bond and forms a strong bond as can be seen if you look at the bond dissociation energies (as always, this is a simplification but it is a useful simplification).
You cannot use a curly arrow to show this as a curly arrow represents the movement of two electrons. Here one electron moves in one direction and the other moves towards the opposite atom. Instead, a half arrow or curly fishhook is used to show that only a single electron is moving. As each bond comprises two electrons, you will need two curly fishhooks every time you make or break a bond.
The next step of the initiation process involves formation of a strong O–H bond at the expense of a weak H–Br bond. The oxygen radical abstracts a hydrogen atom from HBr to form benzoic acid and a bromine radical (or bromine atom). This is the chain carrier. The reaction is similar to deprotonation except that a hydrogen atom transferred (a proton and an electron). Two curly fishhooks form the new O–H bond and one gives the other electron of the H–Br bond to the bromine.
The bromine radical enters the propagation steps. It does the actual chemistry. As you will see, the bromine radical keeps getting regenerated and for this reason it is sometimes called the radical chain carrier.

The second set of steps are the propagation steps. These are the reaction steps that make the product. The chain carrier adds to the alkene to give a carbon-centered radical. The product is formed by hydrogen abstraction. This also regenerates the chain carrier, which then repeats the steps until all starting material is consumed or a termination step occurs.
The propagation steps start with radical addition of the bromine radical to the alkene. This leads to a new carbon-centered radical. The stability of this radical is key to understanding the anti-Markovnikov addition, but I’ll finish the mechanism before discussing regioselectivity. There isn’t a huge driving force for this reaction, the bonds made and broken are of similar strength. The reverse reaction, the elimination is possible but subsequent steps drive the equilibrium forward.
The carbon-centered radical abstracts a hydrogen atom from more HBr. This gives the product and regenerates the bromine radical or chain carrier. This leads to a strong C–H bond at the expense of a weak H–Br bond. The chain carrier can repeat the propagation steps, forming more product. The reaction continues until all starting material is consumed or a termination step occurs.
The termination steps occur when any two radicals meet. They are called a termination reaction as they stop the chain reaction by removing radicals from the propagation step. To minimize termination reactions the concentration of radicals should be kept as low as possible. This is achieved by performing the reactions in dilute solutions and adding the initiator (or one of the reactants) slowly.
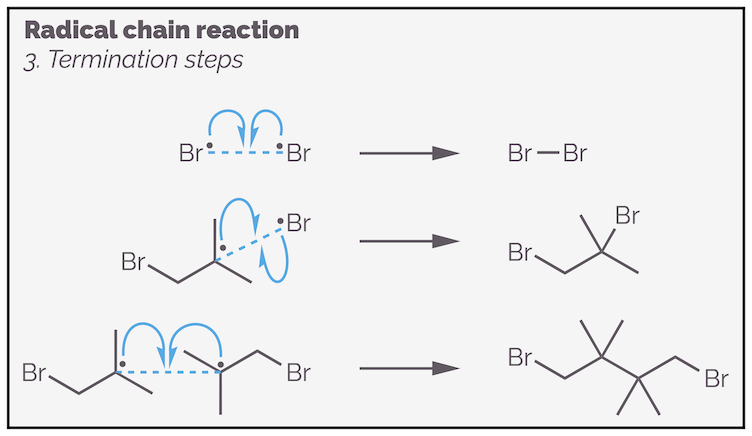
Termination steps remove radicals from the chain reaction. These are just some of the possibilities. The initiator could also be involved.
The overall radical chain reaction can be generalized as:
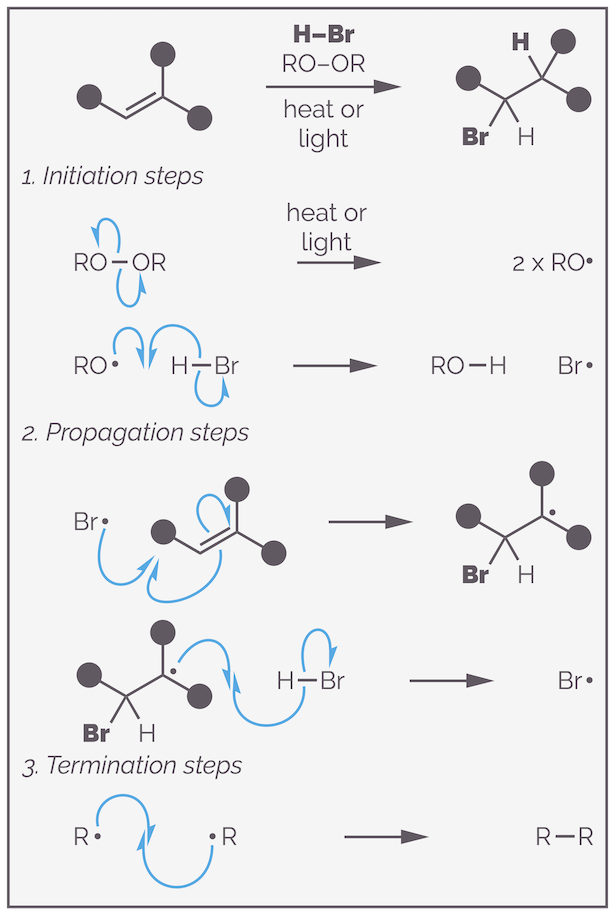
The complete radical chain mechanism for the anti-Markovnikov addition of HBr to an alkene.
Why does the radical addition of HBr reverse regioselectivity and give the anti-Markovnikov product? A carbon-centered radical is electron deficient. It has seven valence electrons instead of a full outer shell of eight electrons. The same factors that determine the relative stability of carbocations determine the stability of a radical. Hyperconjugation, electron donation from simple alkyl groups and resonance delocalization will stabilize a radical.

Factors influencing the stability of radicals, which are electron deficient species.
As C-centered radicals are electron deficient, electron donating groups stabilize them. This means that radicals with more alkyl substituents are more stable than those with fewer. So tertiary radicals are more stable then secondary radicals, which are in turn more stable than primary radicals. Delocalization will also stabilize a radical in the same way it can stabilize a carbocation. Sharing π electrons increases the electron density on the carbon of the radical.
So both Markovnikov and anti-Markovnikov addition are controlled by the same principle, the stabilization of an electron deficient intermediate. The difference is how this intermediate is formed and, ultimately, which atom is added first.
In a radical addition reaction, the bromine radical adds first leading to two possible intermediates:

Addition of the bromine radical can occur to either end of the alkene but favors formation of the more stable tertiary radical intermediate over formation of the less stable primary radical.
Addition of the bromine radical can lead to either a primary or tertiary carbon-centered radical. The tertiary radical has three electron donating alkyl groups stabilizing the electron deficient radical and so its formation is favored over that of the primary radical. The reaction then proceeds as before to give the anti-Markovnikov product.
The ionic addition of HBr proceeds through addition of a proton and formation of the most stable carbocation. Addition of the bromide anion leads to the bromine being on the more substituted carbon or the Markovnikov product. The radical addition starts with the addition of a bromine radical and the formation of the most stable carbon radical. Hydrogen abstraction leads to the least substituted alkyl bromide or the anti-Markovnikov product.
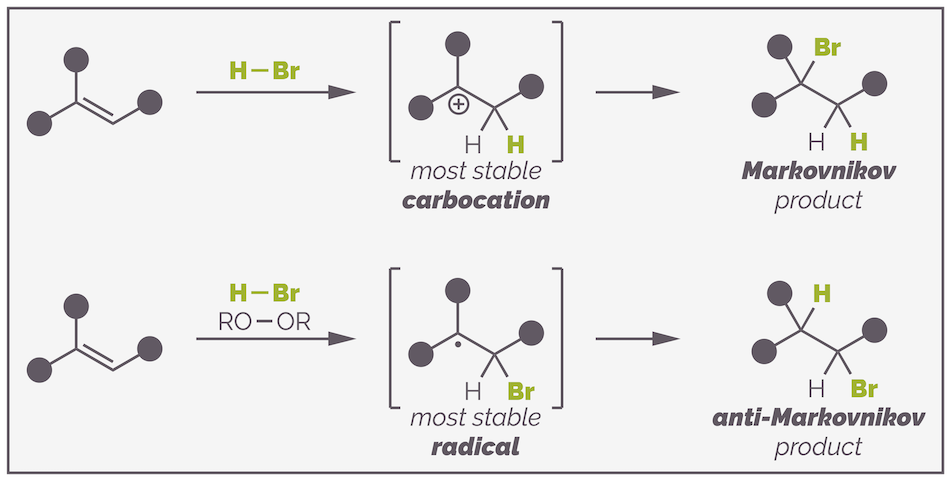
Obtaining either the Markovnikov or anti-Markovnikov product. In both reactions the regioselectivity is determined by the stability of the electron deficient intermediate.
Anti-Markovnikov Addition of Water Across an Alkene
The anti-Markovnikov product of the addition of water to an alkene can be achieved by reacting the alkene with borane, or an organoborane, and then oxidizing the resulting product. This is known as hydroboration-oxidation and is shown for the cyclohexene derivative below:

The hydroboration-oxidation of this cyclohexene derivative gives a racemic mixture of two alcohols, the products of anti-Markovnikov addition.
I’ve used a cyclic alkene in the example above as this highlights the two forms of selectivity that occurring in this reaction. Obviously, the reaction is regioselective and gives the anti-Markovnikov product with the alcohol on the least substituted end of the alkene. The reaction is also stereospecific, the alcohol and the hydrogen atom are both added to the same face of the alkene. This is an example of syn addition. On the left-hand enantiomer, they both added to the bottom face while in the right-hand enantiomer, they both added to the top face. This may be hard to see as I haven’t included the hydrogen or the C–H bond but this is because I’ve drawn the products as an organic chemist would draw them. In the discussion below, I will add the hydrogen to make it clearer. Any mechanism must explain both forms of selectivity.
The first reaction is hydroboration or the addition of B–H across the alkene. This is normally depicted as the reaction of borane, BH3, and an alkene, but this is a simplification. Boron is in group 13. It forms trivalent neutral species. These have only six valence electrons and an empty 2p orbital. They are reactive compounds as they want to gain a full octet and share two more electrons from an appropriate electron donor (Lewis base). Borane itself, BH3, doesn't normally exist (it requires very specific conditions) and is a dimer, B2H6, with a weird three atom-two electron bond. Commonly, borane is used as an adduct with an ethereal (Lewis basic) solvent, tetrahydrofuran (THF) or diethyl ether (Et2O), or the dimethylsulfide (DMS) adduct. Alternatively, bulky organoboranes, can be made or purchased.

Common (organo)boranes used in organic synthesis.
Hydroboration is a concerted reaction, with the C–B and C–H bonds being made at the same time as the alkene π and B–H bonds are broken. The reaction proceeds through a four-membered transition state. This explains the stereospecificity or syn addition of the reaction with both the boron and hydrogen forced to add to the same face of the alkene.
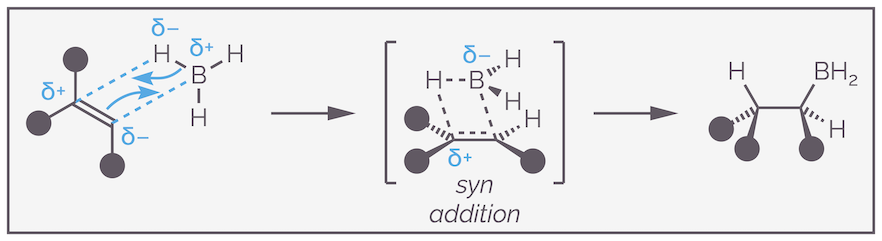
Hydroboration is a concerted reaction that leads to syn addition. The transition state is a four-membered ring that has the boron atom sitting over the alkene.
The regioselectivity can be explained by both electronic and steric arguments. The alkene is the nucleophile, the electron rich π bond attacks the boron. The boron, with its empty 2p orbital, is electron deficient and is the electrophile. The alkene attacks the boron, donating its π electrons into this 2p orbital. This is why I draw the transition state with the boron sitting over the alkene rather than a square four-membered ring (see below for the orbital interactions). As the alkene beginnings to donate its electrons, a partial positive charge builds up. This partial positive charge is more stable on the more substituted end of the alkene. This eventually, leads to anti-Markovnikov addition.
An alternative way to visualize the electronic argument is to think about the polarization of the bonds. Boron is less electronegative than hydrogen (2.04 versus 2.20) and the hydrogen drags electrons towards it becoming more like a hydride, the hydrogen is δ–. The alkene is also polarized. The more substituted carbon has more electron donating groups on it (hyperconjugation). These push the electrons towards the other end of the alkene, making the less substituted carbon partially negatively (δ–) charged and leaving the more substituted end slightly positive (δ+). When the reaction occurs, you want to match the partial charges so that the partial negative of one reactant meets the partial positive of the other. This is shown in the scheme above.
There is also a steric component to the regioselectivity. The larger BH2 will add to the smaller, less sterically demanding end of the alkene, while the small hydride-like hydrogen will add to the larger, more substituted end of the alkene. The steric effect can be enhanced by using a bulky organoborane instead of borane. The reaction of borane with hex-1-ene shows good regioselectivity (94% favoring anti-Markovnikov addition), but this can be increased to more than 99% by using the massive 9-BBN reagent. The rings repel the more substituted end of the alkene.
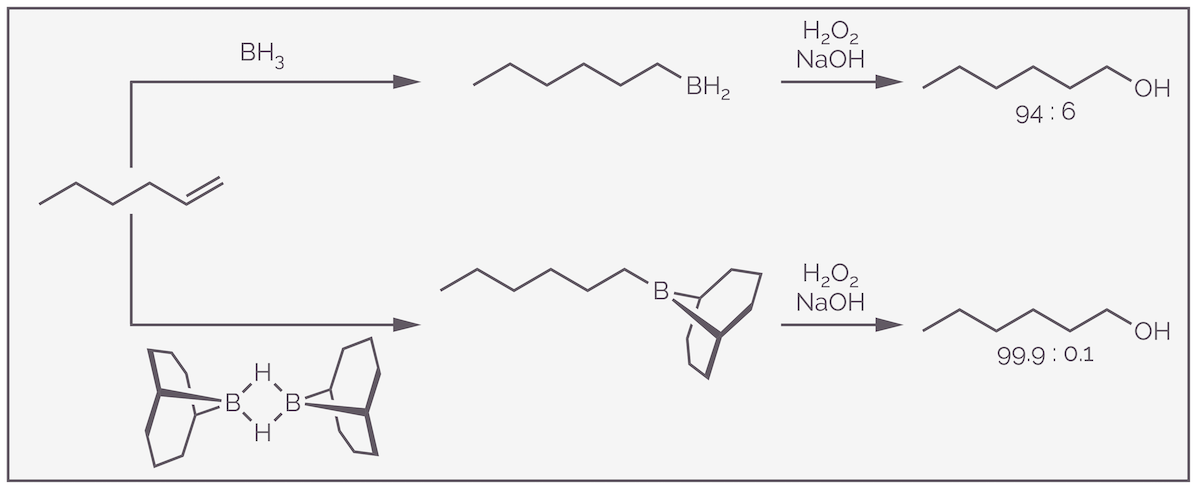
Steric hindrance influences the regioselectivity of hydroboration. The bulkier the boron reagent the greater the preference for the anti-Markovnikov product.
Both the steric and electronic factors favor the boron adding to the less substituted end of the double bond. This eventually leads to the formation of product of anti-Markovnikov addition.
When borane is used in the reaction, the initial product still has two B–H bonds, and it will add to more alkene. Each addition is slower and more selective than the previous. Unhindered alkenes will add three times. Bulkier alkenes might only add once or twice.

Borane can add to three alkenes.
The organoboranes formed by hydroboration are rarely stable. The boron atom still has an empty 2p orbital making it a strong Lewis acid that will react with many electron donors. Reacting the organoborane with a basic solution of hydrogen peroxide (H2O2) leads to oxidation and conversion of the three C–B bonds to C–O bonds. The example below uses the cyclic example again to highlight the stereospecificity:
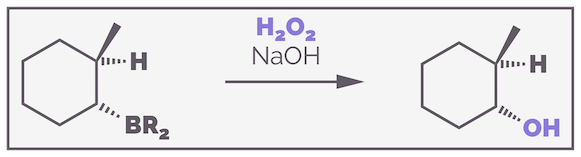
The oxidation of an organoborane with basic hydrogen peroxide is stereospecific, occurring with retention of stereochemistry.
You should notice that the stereochemistry has not changed. The C–B was downwards and the new C–O is also downwards. The oxidation is stereospecific and occurs with retention of stereochemistry.
As before the properties of boron are at the heart of this process. The reaction mechanism starts with the deprotonation of hydrogen peroxide to create a a new nucleophile. This is an example of a proton transfer. The organoborane is a good electrophile due to the empty 2p orbital (the six valence electrons on boron). The hydroperoxide anion adds to the organoborane. I have drawn this with one alkyl substituent and the others represent by R just to make the diagram clearer (it is already big enough!).
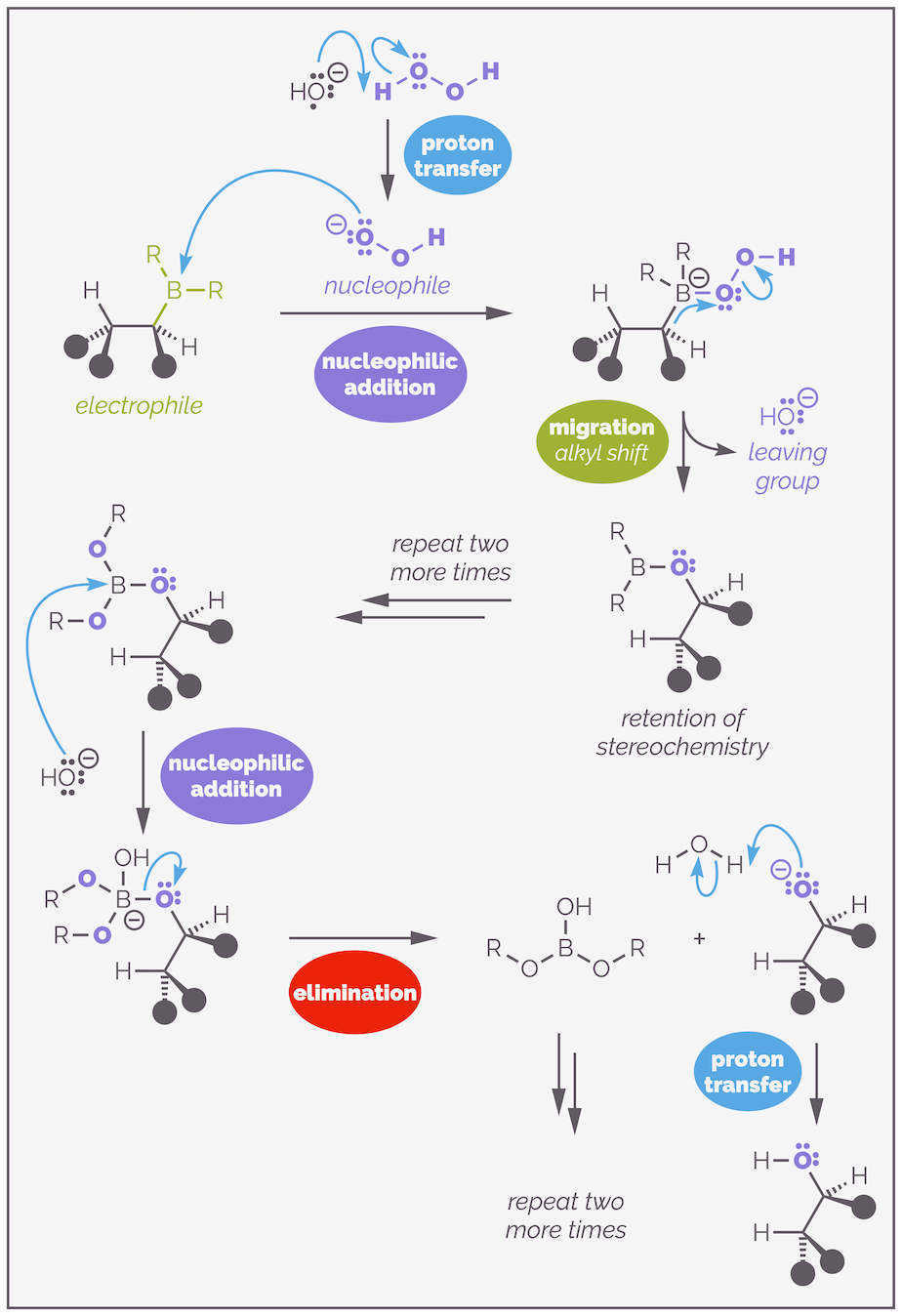
The oxidation of organoboranes occurs with the retention of stereochemistry due to the migration or alkyl shift.
Addition of the hydroperoxide anion leads to a boronate anion. Next is an alkyl shift or alkyl migration. The boron needs to lose the negative charge. It can do this if an alkyl group takes the two electrons of the C–B bond and shifts them to the electronegative oxygen atom. This leads to the weak O–O bond being broken and loss of the hydroxide anion as a leaving group. Key to this reaction is that the C–B bond does not break before the new C–O bond starts to form. This migration occurs through a three-membered transition state where there are partial bonds between the carbon, the boron and the oxygen. I was taught to draw such a shift with the curly arrow ‘upside down’ so that it almost passes through the central atom. This indicates that the carbon is sliding across the atoms rather than breaking one bond and then attacking the oxygen. As there is always some bonding present and not the formation of a carbon centered anion, the stereochemistry does not change or is retained.

The transition state of the alkyl shift or migration shows that there are partial C–B and the C–O bonds. This leads to the retention of stereochemistry. If one bond broke before the other started to form it is likely that there would be lose of stereochemical information.
As there are three C–B bonds, the hydroperoxide anion adds three times, the migration occurs three times and the hydrolysis to give the alcohol and boric acid, B(OH)3, occurs three times.
The hydroboration-oxidation delivers the product of anti-Markovnikov addition of water across the alkene. The reaction occurs with regioselectivity and stereospecificity. The boron adds to the least hindered end of the alkene (anti-Markovnikov). There is the syn-addition of hydrogen and boron so they must be on the same face. The oxidation occurs with retention of stereochemistry so the hydrogen and the oxygen effectively add syn to the alkene.
Taking it further …
The information above is sufficient for most courses but, as always, it is useful to look at the orbital interactions as they give more information about the reaction. The two important steps in hydroboration-oxidation are the hydroboration (surprise) and the alkyl migration during the oxidation.

The orbital interactions involved in hydroboration.
The first interaction in hydroboration leads to the π complex. The π electrons of the alkene are shared with the empty 2p orbital (non-bonding orbital) of the (organo)borane. This can be represented in multiple ways. The three-membered, cyclic, transition state shows the build up of partial positive charge on the carbon atoms that occurs as the electrons are donated. This starts to explain the regioselectivity, with the partial charge on the more substituted carbon being favored due to hyperconjugation. The orbitals show that the (organo)borane will sit above the alkene so that the π bond overlaps the 2p orbital.
The π complex rearranges into a four-membered transition state with the B–H σ bond donating its electrons into the π* antibonding orbital. This leads to the formation of the new C–H bond and breaking the C=C bond. The hydride-like hydrogen (partial negative charge as hydrogen is more electronegative than boron) will match up with the partial positive charge. The transition state is not a square. The boron is moving from the π complex and the ‘hydride' needs to overlap with the π* antibonding orbital that, due to the node, points slightly away. This is important when you consider that hydroboration can be stereoselective as well as stereospecific. The borane will approach alkenes from the least hindered face of the alkene.
These interactions show the alkene forming a C–B bond and the borane forming the C–H bond. These events are concerted (but slightly asynchronous. I’ve drawn one occurring before the other as this is believed to happen) and show that there must be syn addition.
The oxidation occurs with nucleophilic attack of the hydroperoxide anion on the borane. This is simply a lone pair of electrons on the oxygen, which can be considered to be in an sp3 hybridized orbital (but its not important) overlapping with the empty 2p non-bonding orbital of the borane. The interesting interaction is the alkyl shift or migration.
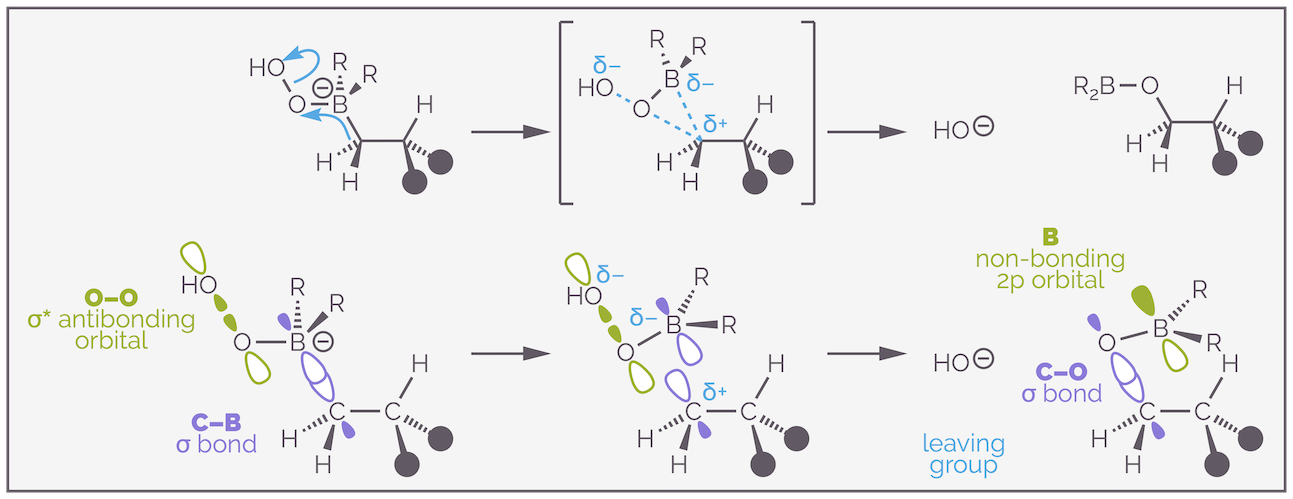
The migration or alkyl shift during oxidation occurs with retention of stereochemistry. Effectively, the C–B σ bond slides across into the O–O σ* antibonding orbital.
The C–B σ bond slides or shifts from the boron to the oxygen. Initially, the electrons are in a σ bond, then they overlap the O–O σ* antibonding orbital. This gives a three-membered transition state with the C–B bond starting to break, the C–O bond starting to form and with electrons entering the antibonding orbital so the O–O bond starts to break. As this is occurring at the same time, no carbanion is formed and stereochemical information is retained.
… end of extra info
Summary
Chemists are always looking for ways to control selectivity. The common addition of electrophiles across a nucleophilic alkene leads to the so-called Markovnikov addition product, where a hydrogen atom adds to the least substituted end of an alkene, and the heteroatom adds to the more substituted carbon. Invariably, this is the result of the reaction favoring the formation of the most stable carbocation or partial positive charge. There are methods to reverse the regioselectivity and form the anti-Markovnikov product. One of the most common methods is the use of radical chemistry to give the anti-Markovnikov addition of H–Br. By forming a radical and adding the bromine atom first, this chemistry leads to bromine being on the least substituted carbon of the alkene and the electron deficient carbon radical stabilized on the more substituted end. Radical addition also introduces the radical chain reaction. Here the radical chain carrier propagates the reaction being involved in the initial addition but also being regenerated on product formation. The second method is hydroboration-oxidation that gives the anti-Markovnikov addition of water across an alkene. This chemistry centres on the properties of boron. Not only is this reaction regioselective, but it is also stereospecific with boron and hydrogen undergoing syn addition to the alkene. Both sterics and electronics control the regioselectivity. The bigger boron adds to the least substituted end of the alkene while the hydrogen is at the most substituted. The two atoms are added to the same face in a concerted reaction. Oxidation of the boron then gives an alcohol, this occurs with retention of stereochemistry.