Conjugate Addition (1,4- or Michael Addition)
Introduction
By now you should be happy that aromatic rings (benzene rings) are normally considered nucleophilic. The delocalized π electrons can be donated to a suitably powerful electrophile to form a new bond. This is electrophilic aromatic substitution where a hydrogen atom is exchanged for something more useful. But the opposite reaction is also possible. Add suitable electron withdrawing groups and the ring becomes electrophilic, it can be attacked by a nucleophile. Now, it is possible to exchange a leaving group in nucleophilic aromatic substitution.

Aromatic rings such as benzene are normally nucleophilic but, by the addition of electron withdrawing groups it is possible to make them into the electrophile and they can be attacked by a suitable nucleophile.
Alkenes are the same. They are normally nucleophilic. Simplistically, you only need two electrons to join the carbon atoms together, and the two electrons of the weak π bond are readily donated to create a new bond. Electrophilic addition reactions were discussed HERE. Like the aromatic systems above, it is possible to reverse the reactivity of alkenes by adding electron withdrawing groups and activating the alkene to nucleophilic attack or making them into electrophiles.

Like aromatic rings, alkenes can be either nucleophiles or electrophiles. Like aromatic rings they are nearly always nucleophiles (even the α,β-unsaturated ester above will react with bromine), but if they have electron withdrawing groups attached they can be electrophiles.
This post is all about electrophilic alkenes and their reaction with nucleophiles. This reaction goes by many names including Michael additions, conjugate additions or 1,4-additions.
Conjugate additions (1,4-additions or Michael additions)
The key to these reactions is activation of the alkene. Once activated a range of nucleophiles will add. Activation requires that the alkene be conjugated with a double or triple bond attached to an electronegative atom. No conjugation, no reaction. Hence one of the names of this process is conjugate additions. In the examples below, the alkene is always in conjugation with a carbonyl C=O double bond. Later I will show you that other functional groups can activate the alkene.
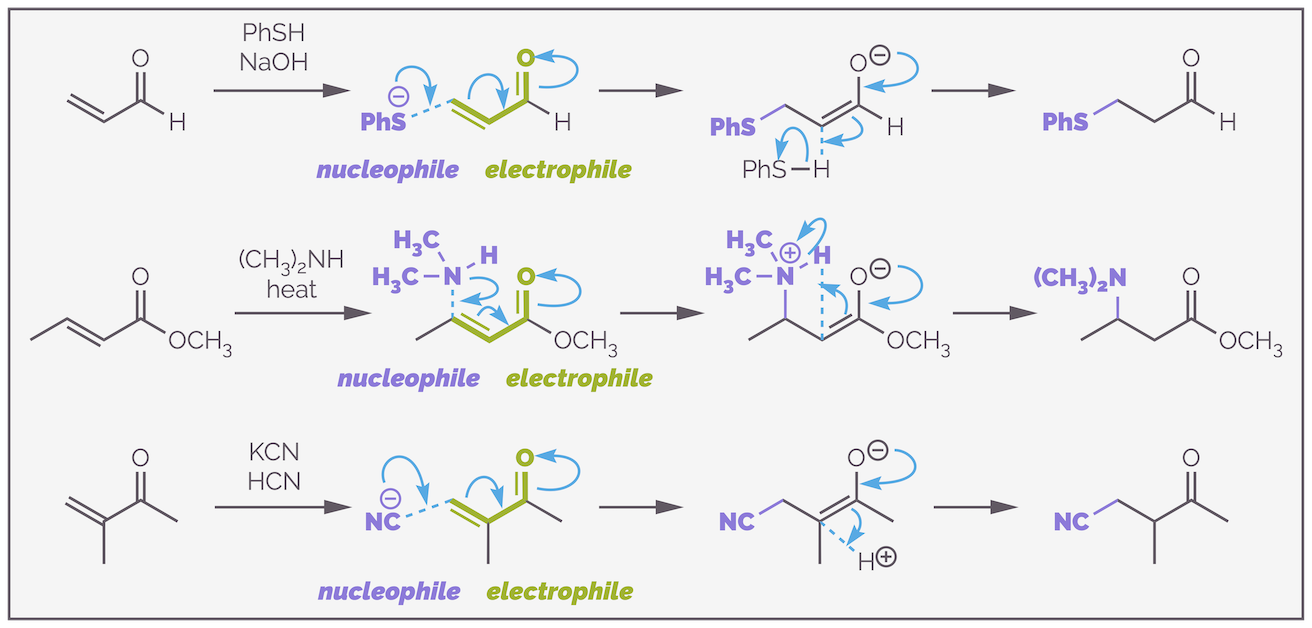
Three examples of Michael additions or conjugate additions or 1,4-additions. The key to each reaction is that the alkene is in conjugation with a carbonyl group.
Conjugated addition results in the nucleophile adding at the carbon of the alkene furthest from the electron withdrawing group (as you shall see, addition can occur elsewhere but the reaction isn't called conjugate addition when that happens!). The nucleophile donates electrons to the substrate and the intermediate will have a negative charge. By adding at the far end this allows the electrons to flow out onto the electronegative atom. The intermediate will be protonated to give the product.
As is always the case in organic chemistry, a lot of terminology has grown up around these reactions. The trivial name for a conjugate additions is Michael addition and the activated alkenes are often called Michael acceptors. Alternatively, the conjugated alkene can be called an α,β-unsaturated aldehydes/ketones/esters. This name refers to the distance from the carbonyl group to the alkene; the first carbon is the α carbon, the second is the β carbon (the third is the γ carbon for those that like their Greek letters). Alternatively, there is a numbering system that starts with the carbonyl oxygen. This leads to the β-carbon or the carbon attacked being known as carbon-4 or the 4 position. Conjugate additions are then termed 1,4-additions.

Conjugated alkenes have a number of different names. They can be called α,β-unsaturated carbonyl compounds as the alkene, the double bond or unsaturated (doesn't have the maximum number of hydrogen atoms possible) is between the α and β carbon atoms. If the alkene was one atom along, a β,γ-unsaturated system it would not reacted in a Michael addition as it would not be conjugated. Alternatively, the atoms can be numbered from the higher priority oxygen. This makes the β-carbon that is attacked by the nucleophile, carbon 4.
Mechanism of conjugate addition
Let's look at the addition of a secondary amine to an α,β-unsaturated carbonyl. The nitrogen is sufficiently nucleophilic that it does not need to be deprotonated prior to attack. It adds to the β-carbon and the electrons flow onto the electronegative oxygen atom. The negative charge is happy here. Such a species is known as an enolate.

Step 1 of the conjugate addition is nucleophilic addition to the activated alkene. This leads to an enolate (or similar species).
The second step is proton transfer to give a neutral species. Proton transfers are fast reactions and there are many different pathways, any of which could be operating (this is why many organic chemists ignore this step or just write +H+). The simplest version of the mechanism has step 2 being an intramolecular (or internal) proton transfer. The carbonyl is reformed, as the C=O double bond is relatively strong and the α-carbon is protonated by the ammonium cation to give the neutral species (top drawing).
An equally acceptable pathway involves two separate intermolecular (external) transfers. First, the ammonium cation is deprotonated by a suitable base (virtually any base is capable of deprotonating the ammonium cation (solvent, amine starting material, amine product)), then the second transfer leads to protonation of the enolate. Here, the conjugate acid could act as the proton source. The scheme below shows the amine starting material as the initial base and its conjugate acid as the proton source (bottom drawing).

The second step of conjugate addition involves proton transfer. In this example this is required to deprotonate the ammonium cation and protonate the enolate. In many examples, step 2 is simply the protonation of the enolate.
There is a third set of allowable proton transfers. Instead of protonating the β-carbon of the enolate, it is possible to protonate the oxygen to give an enol. This will rearrange to regenerate the stronger carbonyl at the expense of the weaker C=C in a process called tautomerization. Tautomerization is the fancy term for shifting a proton (and normally a double bond) within a molecule. This process is more likely when there are two activating groups as such found in the product of the Knoevenagel condensation (HERE).
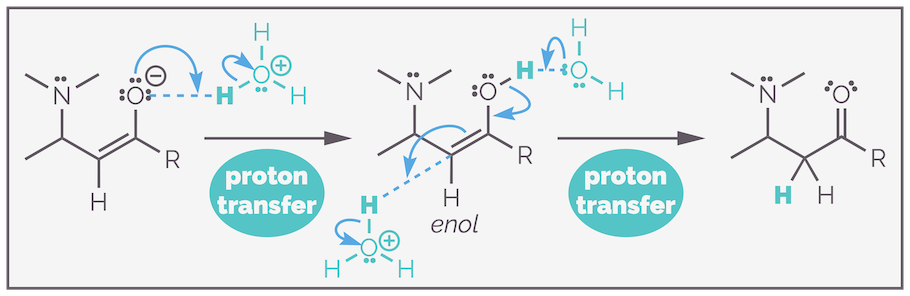
Yet another mechanism to form the product involves protonation of the oxygen to give an enol. The enol undergoes tautomerization (and let's be honest, there are multiple mechanisms for this process as well) to give the product. This just adds extra steps and seems totally unnecessary unless ...
To be brutally honest, the exact order of these proton transfers and whether they are inter- or intramolecular is not very important. All of these mechanisms are allowable and all probably occur.
There is one mechanism when the order of proton transfer is important, and that is if the reaction is acid catalyzed. In which case, proton transfer occurs prior to the addition and the mechanism can be drawn as below:

Some conjugate additions can be accelerated by the addition of acid. This leads to protonation of the electrophile with the new, cationic species, being more electrophilic. Addition occurs as normal to give the enol directly. A series of proton transfers cause tautomerization and the formation of the product.
Why does the nucleophile add to the β-carbon and not the α-carbon of the conjugated alkene? An easy explanation involves the resonance structures and shows that the alkene is polarized.
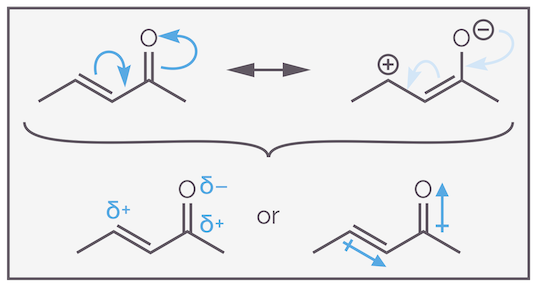
The resonance structures of a typical enone show why the alkene is polarized and activated towards nucleophilic attack at the β-carbon (4 position).
The electrons flow towards the electronegative oxygen, and this reveals that one of the resonance structures has a positive charge on the β-carbon. This means the alkene is polarized in such a way that the nucleophile is attracted to the β-carbon.
This section has covered a lot of ground, and possibly over complicated the reaction! But I have tried to address the most common questions students have (alright, the most common question is actually in the next section).
This section can be summarized by the general mechanism below, which shows the conjugate addition of a generic nucleophile to a carbonyl-based activated alkene:

The general mechanism for a conjugate addition involves two steps. The first is addition of the nucleophile to form an enolate. The second step is a proton transfer that returns a neutral species with the strong carbonyl group intact.
Chemoselectivity
There are two electrophilic atoms within the carbonyl-containing molecules above and this leads to the issue of chemoselectivity, which functional group will react, the alkene or carbonyl group? The electronegativity of the oxygen makes both the carbonyl carbon and the alkene carbon electrophilic. This means there are two sites a nucleophile can attack. Below, I've shown the resonance structures again and the two possible modes of attack:
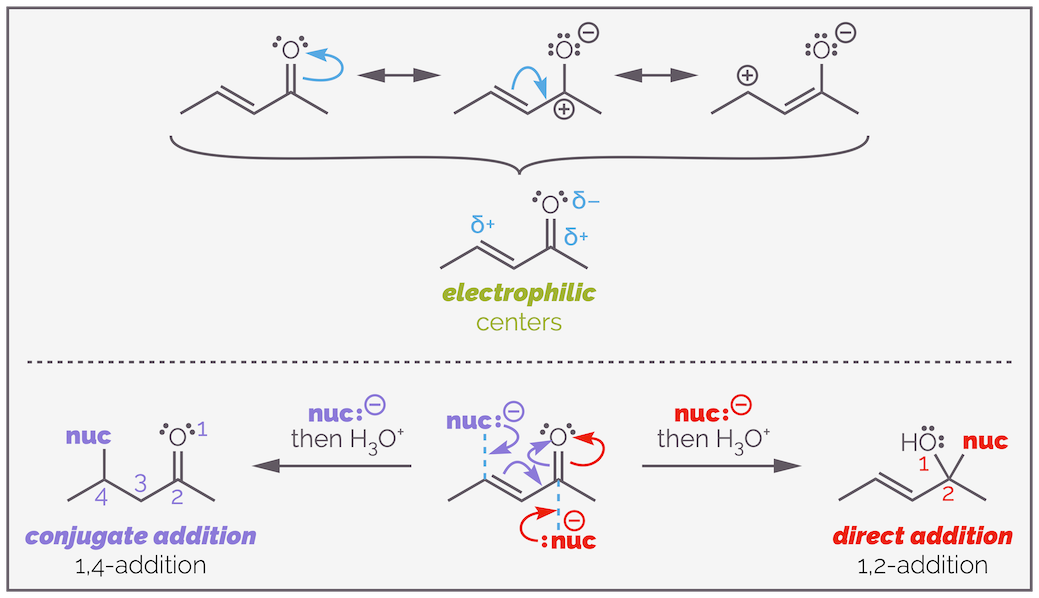
The conjugated carbonyl group polarizes the alkene and makes it electrophilic but it also adds a second electrophilic center to the molecule. The resonance structures show that the molecule has two electrophilic centers, the carbon of the carbonyl group (carbon 2) and the β-carbon of the alkene (carbon 4). A nucleophile can add directly to the carbonyl group to give the product of 1,2-addition or it can participate in conjugate addition to give the 1,4-addition product.
Michael addition, or conjugate addition or 1,4-addition occurs at the alkene (shock horror considering the section above). Direct addition occurs directly to the carbonyl group and is sometimes called 1,2-addition. Where the nucleophile attacks is controlled by type of nucleophile, the structure of the electrophile and the reaction conditions. This blog post is not meant to be a comprehensive dive into the chemistry of conjugate additions but a summary, and so I will focus on the type of nucleophile (but will mention other factors in passing).
There are a number of ways of rationalizing the chemoselectivity of various nucleophiles and I will discuss the two most common arguments. To understand the first it is necessary to understand the difference between the kinetic product and the thermodynamic product.
The thermodynamic product is the most stable product. It is the compound with the lowest energy. The kinetic product is the compound that is formed the quickest. It has the lower activation barrier, in other words, the transition state leading to this product is lower in energy than the transition states leading to other compounds. This is shown in cartoon form in the diagram below:

A cartoon plot showing the difference between the kinetic and thermodynamic product.
If the imaginary reaction above is performed at low temperature it can be assumed to be irreversible. This means the predominant product will be the kinetic product, as this is formed first (or formed fastest). If the reaction is performed at high temperature then there can be sufficient energy to make the processes reversible. The kinetic product is still formed first, but now it can return to starting material, and over time more reactant is converted to the stable thermodynamic product. The reverse reaction is harder as there is a bigger activation barrier to the backwards process (the compound is more stable). This means the longer the reaction is run, the more thermodynamic product is formed.
Simplistically, if the reaction is irreversible or left for a short period of time, the kinetic product is formed. If the reaction is reversible and left for a long period of time, the thermodynamic product is favored.
Before going any further, you should note that while the kinetic and thermodynamic products can be different, there is no reason they have to be. In some reactions the thermodynamic product is also the kinetic product. In such reactions, selectivity isn't an issue.
Why is this important? Nucleophilic attack on the carbonyl group is fast. The carbonyl bond is highly polarized making the carbon very electrophilic. Attack on the alkene is considerably slower. The alkene is polarized but to a far less extent. It is further from the electronegative oxygen atom and the effects are less. Attacking the carbonyl group is the kinetically favored reaction.
With the correct nucleophile, one that is a weak base (it has a strong conjugate acid with a low pKa), addition to the carbonyl group will be reversible. The reactions of carbonyl-containing functional groups (HERE, HERE, HERE & HERE), are dominated by collapse of the so-called tetrahedral intermediate to reform the strong carbonyl group. If there is a leaving group (a weak base) then additions to the carbonyl group are reversible. Attack at an alkene tends to be irreversible. Attack at the alkene also leads to the thermodynamically favored product. Alkene C=C bonds are weaker than carbonyl C=O bonds. The product that retains the C=O bond at the expense of the weaker C=C bond is more stable and favored.
All this can be seen if you look at the reaction of cyanide with an enone:
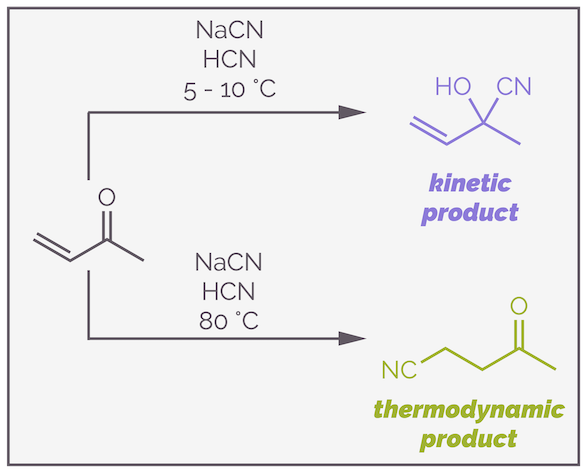
At low temperature the addition of cyanide to an enone favors formation of the cyanohydrin. At higher temperatures, the initial reaction becomes reversible and the thermodynamically more favored molecule becomes the predominate product.
The key to the change in reactivity is the potential reversibility of cyanohydrin formation. The cyanide anion reacts more rapidly at the ketone. This is the kinetic product. At low temperature, there is insufficient energy for the reverse process but if the reaction is warmed, then formation of the cyanohydrin becomes reversible. Now, some of the reagents react through 1,4-addition to give the thermodynamic product in an irreversible process. This removes one side of the equilibrium (HERE & HERE) and slowly, over time, the reagents are funneled down this pathway until the majority of the product is the Michael addition product.

Heat the reaction of an enone with the cyanide anion and there is sufficient energy to make cyanohydrin formation reversible. Now the slow, but irreversible 1,4-addition becomes more important. Given sufficient time, the majority of compound will be syphoned down this pathway to give the thermodynamic product.
If a nucleophile is a strong base (it has a weak conjugate acid with a high pKa) then it adds to the carbonyl group irreversibly. A strong base is a poor leaving group and re-formation of the carbonyl group is disfavored. This means you will observe the direct or 1,2-addition of the nucleophile. A good example of this is the use of organolithium reagents such as butyllithium. This is shown below:

A nucleophile that is a strong base (it has a weak conjugate acid) will undergo irreversible direct addition to the carbonyl group or 1,2-addition. This is an example of a reaction under kinetic control.
If the nucleophile is a weak base (it has a relatively strong conjugate acid with a low pKa), then addition to the carbonyl group is often reversible. Species that are weak bases are often good leaving groups. The reaction is now under thermodynamic control and conjugate, or 1,4-addition is favored. This keeps the strong carbonyl at the expense of the weaker alkene. Heteroatomic nucleophiles tend to fall into this category. For example, alcohols predominantly give the 1,4-product.
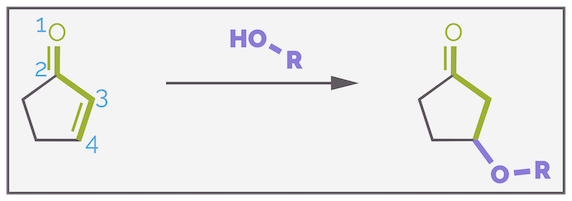
The addition of alcohols, which are weak bases, predominantly gives the product of 1,4-addition.
Some carbon-based nucleophiles can participate in conjugate or 1,4-additions. You have already met one, the cyanide anion. It is a weak base or is a relatively stable anion. It adds reversibly to the carbonyl group. Other examples are also weak bases that have a stabilized carbanion. The most common are examples are di-carbonyl compounds such as the β-ketoester below. The enolate anion is delocalized over both carbonyl groups, and is relatively stable. This leads to reversible addition to the ketone, allowing reactions to be performed under thermodynamic control. This leads to 1,4-addition.

The conjugate addition of a resonance stabilized nucleophile.
The second argument that explains why some nucleophiles add directly while others undergo conjugate addition involves considering hard/soft nucleophiles/electrophiles. Hard nucleophiles tend to have a negative charge concentrated on a small, electronegative atom. The reactivity of hard nucleophiles is dominated by electrostatic forces. Soft nucleophiles are normally large and diffuse, with any charge spread out over a larger volume. Their chemistry is controlled by orbital interactions. Electrophiles can also be viewed as hard or soft. Again, hard electrophiles are small, concentrated areas of charge while soft electrophiles are bigger, more diffuse, and frequently neutral.
Hard nucleophiles react with hard electrophiles, with a strong electrostatic attraction between the concentrated volumes of charge. Soft nucleophiles react with soft electrophiles as there is good overlap between the large, diffuse orbitals.
It turns out that our conjugated system is both a hard and a soft electrophile. The carbonyl is a hard electrophile. The polarization of the C=O bond means there is high partial positive charge on carbon-2. The alkene is polarized by resonance. The electrons are spread out. The build up of positive charge is weaker and more diffuse. The alkene is a soft electrophile. Hard nucleophiles directly attack the carbonyl. Soft nucleophiles attack the alkene.
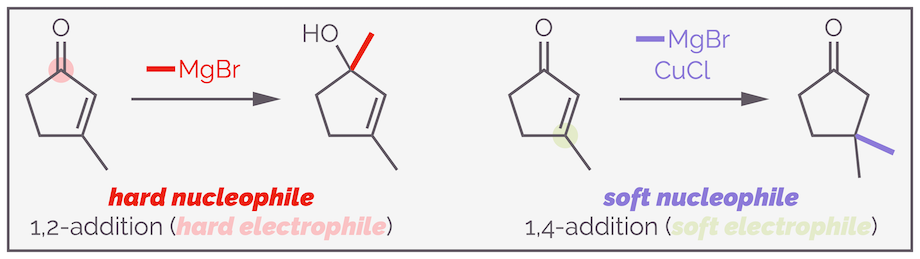
A Grignard reagent is a hard nucleophile and it is involved in 1,2-addition to give an alcohol. Add copper to the reaction and you create an organocopper reagent. These are soft nucleophiles and they participate in conjugate addition.
Grignard reagents (organomagnesium reagents) are hard nucleophiles. The C–Mg bond is highly polarized leading to a build up of negative charge on the carbon. This matches the high build up of positive charge on the polarized carbonyl and Grignard reagents directly attack in 1,2-addition. Add copper and you observe 1,4-addition. The copper transmetalates the Grignard reagent to give an organocopper reagent. These are soft nucleophiles. The large copper is less electronegative than magnesium so causes the bond to be less polarized. The carbon is less partially negative and now reacts with the soft 4-position.
By now, it should be clear that the structure of the nucleophile influences the selectivity between 1,2- and 1,4-addition. The electrophile can also influence reactivity. Different carbonyl-containing functional groups have different reactivities. If you make the carbonyl more reactive by using an aldehyde, then you will increase the possibility of 1,2-addition. Reduce the reactivity of the carbonyl group by adding a heteroatom that can delocalize its lone pair, such as an amide, and you will reduce the polarization of the C=O bond. Now attack at the alkene becomes more likely.
Below is the addition of butyllithium to a conjugated aldehyde and to a conjugated amide. The aldehyde is more reactive and direct addition to form an alcohol is observed. The amide is less reactive and more of the product of 1,4-addition is isolated.
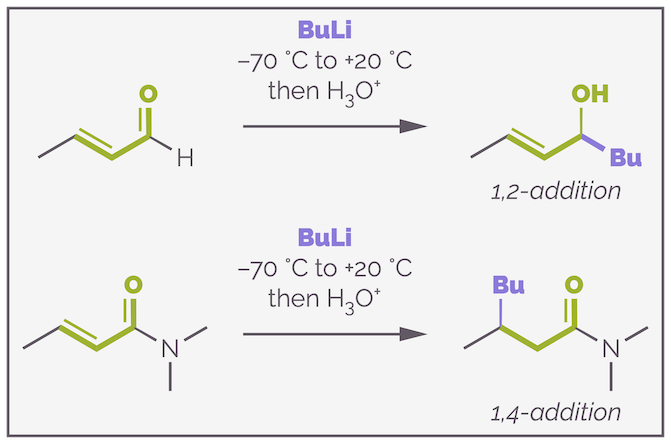
Changing the carbonyl-containing functional group can alter the 1,2- versus 1,4-addition selectivity. More reactive carbonyl groups, like aldehydes encourage 1,2-addition while less reactive carboxylic acid derivatives, such as amides, encourage 1,4-addition.
Other electrophiles
Conjugate additions don't just occur with carbonyl-containing compounds. Any electron withdrawing functional group that is in conjugation with the alkene can make the alkene into an electrophile. Other functional groups have the added advantage that they are frequently not electrophiles themselves (for example, nitro groups) and 1,2-addition is no longer an issue.
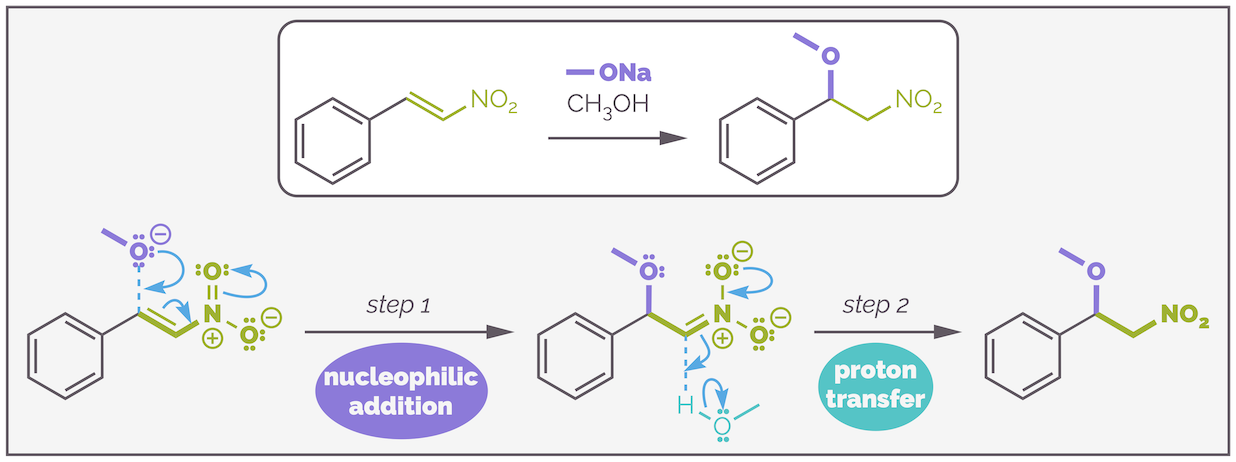
An example of the conjugate addition of a nucleophilic to a nitroalkene. The mechanism is the same as the addition to an enone.
The scheme above shows the addition of an alkoxide (oxygen nucleophile) to a nitroalkene. This another example of a conjugate addition as the alkene is electrophilic due to conjugation to a powerful electron withdrawing group in the form of a nitro grouo. In fact, the nitro group is more electron withdrawing than a carbonyl group (consider our previous discussions in nucleophilic aromatic substitution, a mechanism that is remarkably similar to this one) and the reaction is faster, and there is no opportunity for 1,2-addition. The mechanism is identical to that of the carbonyl activated alkenes. The first step is nucleophilic addition with the electrons pushed out onto the oxygen. This gives the nitro equivalent of an enolate (which is many names including the aci form, a nitronate or an azinate). In the same way that the enolate was protonated, so is the nitro intermediate.
As you saw with the carbonyl-activated alkenes, drawing the resonance structures of the alkene reveals that the β-carbon is the electrophilic center (if pushing the curly arrows above didn't already show you why the nucleophile must attack the β carbon). The electrons are pulled towards the electronegative oxygen atom.

Resonance structures of a nitroalkene showing that the β-carbon is the electrophilic carbon due to the π electrons being dragged towards the electronegative oxygen atoms.
I can draw the same mechanism for any electron-withdrawing functional group that is in conjugation with alkene. The next example is acrylonitrile. It behaves in exactly the same manner as the carbonyl-containing compounds and the nitroalkene as shown by the curly arrows.
No care is required in this reaction as the nitrile group is similar to the nitro group in that it is a poor electrophile itself and the addition is exclusively at the β-carbon (nitrile groups can be attacked but it is very rare). The reaction has the same two steps as before; nucleophilic addition followed by proton transfer (which is probably intermolecular but has been drawn as an intramolecular process for convenience (yes, I'm being lazy)).

The addition of a nucleophilic amine to an electrophilic alkene activated by being in conjugation with a nitrile group.
Another example is the reaction of a sulfone. I don't know why I've added it, as it doesn't tell you anything you shouldn't already been able to work out. Perhaps I just like sulfones (I do). The mechanism is identical to before.

The addition of a nucleophilic thiol to an electrophilic vinylsulfone.
An Aside: An orbital view
The discussion above is sufficient for most undergraduate courses but some might like a little more detail so here is a very simplistic look at the frontier orbitals involved in conjugate addition to an enal.
Any discussion of the important orbitals of an enal tends to start by discussing 1,3-butadiene. The π-system of this is easy to picture as it is formed from the overlap of four 2p orbitals. The changes in phase of the four orbitals (and a rough approximation of the size of the coefficient) can be predicted by considering an electron in a box with an increasing number of nodes. This is shown below. The two most important orbitals are the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).

The π orbitals for 1,3-butadiene. On the left is the 'electron in a box' representation that shows the wavefuntion. In the middle is the more 'normal' representation used by organic chemists then there is a 'top down' view that tries to show the relative size of the coefficient on each atom.
The π-system of the enal is similar. There are the same combinations of 2p orbitals but the presence of the oxygen distorts the size of the 2p orbitals or more accurately, the size of the coefficients on each atom. I don’t want to get into how the size can be calculated, suffice to say the picture below gives a rough idea. Note, it doesn’t say HOMO next the full π orbitals of the enal as the π system isn’t the highest occupied orbital. The lone pair of electrons (or the non-bonding electrons) are higher in energy.

The frontier molecular orbitals of an enal are very similar to those of 1,3-butadiene (except the HOMO is now the non-bonding lone pairs of electrons and are not shown in this diagram). The other difference is that the polarization of the bond changes the size of the coefficients of the orbitals (a good background to this can be found in Ian Fleming’s excellent textbook).
For addition to occur, the HOMO of the nucleophile attacks, overlaps or fills the LUMO of the electrophilic enal. Attack occurs where there is the best overlap of orbitals, and by best, I mean the coefficients of the overlapping orbitals are most similar in size. The two biggest coefficients for the LUMO of enal are on carbon 2 & 4, or correspond to direct attack on the carbonyl versus conjugate addition at the β-position. The biggest coefficient is on the β-carbon. This means a nucleophile with a large coefficient, a nucleophile with electrons spread out or, in other words, a soft nucleophile will attack at the β-carbon. A nucleophile with a more concentrated, smaller, denser charge, a hard nucleophile will attack the carbon with the smaller coefficient. A hard nucleophile directly attacks the carbonyl.
For our first example (somewhere near the top of the page), the HOMO on the thiolate is a lone pair on the sulfur atom. This overlaps with the largest coefficient of the LUMO of enal or it overlaps with the empty orbital on the β-carbon. This overlap leads to a new σ bond.

The HOMO of the nucleophile will overlap with the LUMO of the electrophile. Soft nucleophiles will have a large coefficient on their HOMO as the charge is diffuse, this will overlap with the carbon with the largest coefficient in the LUMO of the Michael acceptor. This is on the β-carbon (carbon 4). Hard nucleophiles, with a smaller coefficient will overlap the slightly smaller coefficient on the carbon of the carbonyl group.
Summary
Conjugate additions occur when an alkene is activated by an electron withdrawing group. Each of these so-called Michael acceptors has a functional group such as a nitro group, a nitrile, a sulfone or a carbonyl-containing group in conjugation with the alkene.

Examples of Michael acceptors. These are conjugated alkenes.
The nucleophiles in these reactions tend to be weaker bases. These include heteroatomic nucleophiles such as amines, alcohols and thiols as well as the cyanide anion, malonates and other β-carbonyl enolates. Organocopper reagents are good nucleophiles in these reactions. Other organometallic reagents tend to add directly to the carbonyl group rather than the alkene.

Examples of nucleophiles for conjugate additions.
Conclusion
Conjugate addition reactions involve the addition of a nucleophile to an activated alkene. In many respects, these reactions are related to nucleophilic aromatic substitution in that a normally nucleophilic group (alkene or aromatic ring) is transformed into an electrophile by the addition of a conjugated electron withdrawing group. This group polarized the alkene and allows the additional electrons gained during nucleophilic addition to flow out onto an electronegative atom. Good activating groups include aldehydes, ketones, esters, amides, nitro groups, nitriles, and sulfones. Alkenes conjugated to these groups are often known as Michael acceptors.
When the electron withdrawing activating group contains a carbonyl group there is an added complication as there are two potential electrophilic centres within the acceptor. Nucleophiles could add directly to the carbonyl group (1,2-addition) or to the β-carbon of the alkene (1,4-addition). Direct, 1,2-addition is kinetically favored or is normally the faster reaction but 1,4-addition is often thermodynamically favored. As long as the reaction is reversible, it can be driven towards the desired product. The choice of nucleophile can also make a big difference to which addition occurs. Nucleophiles that have a strong conjugate base, or have a small, concentrated charge, are known as hard nucleophiles and they favor direct addition. This includes organometallic reagents such as organolithium or Grignard reagents. Nucleophiles with weak conjugate bases or those that have a diffuse charge are soft nucleophiles. These favor the 1,4-addition and include heteroatoms and organocopper compounds.