Electrophilic Aromatic Substitution
Introduction
Alkenes can make good nucleophiles, and I have written a number of posts on electrophilic addition to alkenes. A number of reactions were introduced including bromination, epoxidation, hydrohalogenation and hydroboration. All of these reactions involved the electrons of the alkene attacking a suitable electrophile. This can be exemplified by the bromination of an alkene where the alkene attacks bromine to give a brominium ion that this then attacked by bromide:

The stepwise bromination of an alkene involves the nucleophilic alkene attacking bromine to give a bromonium ion. This is attacked by the bromide anion to give the product of anti addition. All atoms of the starting materials are included in the product.
At first glance, you may think that the archetypal aromatic ring, benzene, would make a good substrate for the bromination reaction. The commonly drawn Lewis structure (one of its resonance structures) of benzene appears to be electron rich with three 'π bonds'. It looks like it should be a good nucleophile and yet benzene does not react with bromine.

Benzene does not react with bromine (on its own). Again, this shows why you must be careful when you look at Lewis structures (or resonance structures), they do not always tell the whole story.
The delocalization of electrons in an aromatic ring leads to increased stability when compared with isolated π bonds. Simplistically, by being able to draw multiple resonance structures for benzene, you can show that the electrons are spread out in a ring and this results in them being lower in energy or more stable. A more stable compound is a less reactive compound. Benzene is not sufficiently nucleophilic to react with bromine.
It is possible to force benzene to react with bromine. This is achieved by activating the electrophile. If you make the electrophile more reactive then it can be attacked by the weakly nucleophilic benzene. This can be achieved by adding a Lewis acid.

The bromination of benzene in the presence of a Lewis acid occurs to give the product of a substitution reaction not an addition (atoms are exchanged between the reactants).
Under these new conditions, benzene reacts with bromine but in a different reaction to the alkene. Benzene participates in a substitution reaction, with a bromine atom replacing a hydrogen atom. It doesn't undergo addition to give a dibromide like the alkene did. Again, the difference can be explained by aromaticity.
This post is all about electrophilic aromatic substitution. I'll discuss the general mechanism, show how different electrophiles can be activated, and then look at the effect of substituents on the reactivity of the benzene ring.
The Bromination of Benzene
The bromination of benzene is a substitution reaction with one hydrogen on benzene being replaced by a bromine atom. The overall reaction is shown below with the appropriate atoms highlighted:

Highlighting the substitution of a hydrogen atom by bromine during electrophilic aromatic substitution.
The success of this reaction is due to the formation of a powerful electrophile through the interaction of bromine with an appropriate Lewis acid. The most common example in textbooks is aluminium trichloride (or the tribromide) but other Lewis acids work equally well. Those of us who work with [2.2]paracyclophane invariably perform brominations by using iron(III) bromide made in situ (in the reaction vessel) from iron and bromine.
The activation of bromine by a Lewis acid is shown below. In many textbooks, it is simplified to the formation of a bromine cation Br+ but it is far more probable that the active species is the Lewis acid complex highlighted in the scheme.

Step 0 - The activation of bromine with a Lewis acid (AlBr3).
You can think of this activation step as being Step 0 (of the general electrophilic aromatic substitution reaction SEAr as it occurs before the aromatic ring is involved). This creates a powerful electrophile that permits Step 1 (normally the rate determining step) to occur. The electrophile is sufficiently reactive that it can be attack by the relatively stable benzene ring. Step 1 is normally called nucleophilic addition as the two reactants add to give a reactive cationic intermediate. This can be confusing as it is arguably a substitution step as AlBr4– is kicked out. This is probably why some textbooks use the bromine cation as it simplifies the mechanism.

Step 1 - Nucleophilic addition. The nucleophilic aromatic ring attacks the activated electrophile to form a cationic intermediate.
Arguably, there is an important step prior to the nucleophilic addition and this is association of the nucleophile and electrophile prior to reaction. This is known as formation of the π complex as it involves the π electrons of the aromatic ring and the σ* antibonding orbital of the electrophile. This interaction is important in explaining regiochemistry but is ignored in most (if not all) undergraduate courses.
The addition step is the slow, rate determining step. It breaks the aromatic ring and leads to the lose of stability that aromaticity accords a molecule. Breaking the aromatic ring takes energy and is the reason you must use activated electrophiles. It is hard.
The cationic intermediate is unstable compared to the starting materials (and the product). It is not aromatic. There is an sp3 hybridized carbon that breaks the ring of p orbitals that is necessary to create an unbroken ring of π electrons. Most chemists include the hydrogen atom at this position to emphasize the change in hybridization and the break in aromaticity.
The intermediate is stabilized by the delocalization of electrons. You can draw a number of resonance structures. These show that the carbocation is spread out over five atoms and can be drawn as the resonance hybrid below. This intermediate is known as the arenium (ion) intermediate or Wheland intermediate or the sigma (σ) complex. It is more useful to concentrate on the resonance structures, as these reveal that the positive charge is focused on three of the five atoms. This will be very important when you start to determine regioselectivity during the reaction of substituted benzene rings.

The arenium ion, σ complex or Wheland intermediate. This is not aromatic due to the sp3 carbon. The charge is stabilized by delocalization but you should remember that it is focused at the three carbons indicated in the resonance structures rather than just spread everywhere (the resonance hybrid).
Step 2 is deprotonation or re-aromatization of the aromatic ring. Aromaticity instils stability in a molecule, and re-gaining aromaticity is a favorable process that drives the reaction forward. Many textbooks show Br– acting as a base and removing the proton. The problem with this is that there isn’t bromide ion floating about and even if there was, it isn’t a good base (the electrons are spread over a large atom. HBr is a strong acid with a pKa of –9). Instead, you should use the aluminium tetrabromide as shown below (to be honest, a really weak base should be sufficient to deprotonate the cationic intermediate so perhaps using Br– is fine!).

Step 2 - deprotonation (or proton transfer) removes a proton and re-forms the aromatic ring.
The reaction regenerates the aromatic ring and the Lewis acid. In theory, the Lewis acid is a catalyst as it promotes the reaction before being re-formed. In practice, it is very rare for sub-stoichiometric (less than one equivalent) of Lewis acid to be used.
The overall mechanism for bromination is shown below. It is three steps, activation, addition and proton transfer (deprotonation). Steps 1 and 2 comprise the reaction known as electrophilic aromatic substitution (SEAr) although I like to include the first step as well as activation is vital in many examples (but not all and that is why it is not an official part of the process).

The complete mechanism for the bromination of benzene. There are three steps: Step 0 is activation of bromine, the electrophile. Steps 1 & 2 are electrophilic aromatic substitution (SeAr) with Step 1 being nucleophilic addition to give the cationic intermediate. Step 2 is deprotonation or proton transfer, which returns the aromatic ring. Overall, a bromine atom has substituted a hydrogen atom.
I know some of you like to take the reactions a little further and discuss the frontier molecular orbitals (or the valence bond model) so here it is:

The frontier orbitals (or valence bond model) for the bromination of benzene. This shows the formation of the π-complex followed by the addition step that leads to breaking of aromatic ring and the synthesis of the cationic σ-complex. The lowest drawing shows proton transfer to reform the aromatic ring and lead to the product of substitution.
First, there is an interaction between the electrophile and the π electrons. This is the so-called π complex. In substituted aromatic rings, the coefficient of the various p orbitals is different, there is greater electron density on some atoms and reduced electrons density on others. This means the electrophile will be more closely associated with specific atoms in this complex. This is probably important in influencing regiochemistry.
The π electrons of the ring are in the highest occupied molecular orbital (HOMO) and will attack the σ* antibonding orbital of the electrophile or lowest unoccupied molecular orbital (LUMO). This leads to the creation of a new σ bond. It is at this point that the hybridization of a carbon atom changes from sp2 to sp3 and the aromatic ring is broken. With the change in hybridization, there is no longer an a p-orbital on the carbon and the ring of π electrons (which was made from a ring of parallel p orbitals) has been broken.
Deprotonation or proton transfer involves the base donating two electrons into the C–H σ* antibonding orbital. This breaks the C–H bond and the carbon changes hybridization becoming sp2. The electrons are now in a p orbital and the unbroken ring of π electrons has been regenerated. The molecule is aromatic once more.
Substituent Effects - Activation, Deactivation and Directing Effects
What happens to bromination if there is already a substituent on benzene (it isn’t benzene anymore!). The addition of a substituent on the benzene ring has two effects, altering the reactivity, either activating or deactivating the ring to addition, and controlling which atom or position around the ring reacts (regioselectivity).
To describe the reactivity, you need to know how the atoms of an aromatic ring are named. It turns out that there are two ways to name or number the positions relative to the substituent. The first is based on the standard name of the molecule and will number the carbons from 1 to 6, starting with the highest priority substituent. The second is far more common and this names the positions relative to the original substituent as shown below:

Naming the positions around a benzene ring relative to a substituent. The positions are either given the names ortho, meta, and para, or are numbered (given in blue inside the ring with the original substituent being position 1 and then other substituents having the lowest possible numbers).
The two positions next to the substituent are the ortho positions. Then there are two meta positions and finally, the position directly opposite the substituent is called the para position.
So far, I have stressed that benzene will only react with an activated electrophiles, yet phenol, a benzene ring with a hydroxyl substituent, is very different, it reacts with bromine at room temperature in the absence of a Lewis acid. What has changed?

The bromination of phenol occurs at room temperature without the addition of a Lewis acid. It is virtually impossible to stop multiple additions at this temperature.
Phenol is more reactive a lone pair of electrons on the oxygen is delocalized into the benzene ring. This donation, or sharing of electrons makes the aromatic ring more electron rich. This makes the ring more nucleophilic and capable of reacting with the weak electrophile, bromine.
The nucleophile is activated and electrophilic aromatic substitution proceeds without the extra step of activating the electrophile. You can draw the curly arrows for this reaction. The key is that they will start with the non-bonding lone pair on the oxygen and reaction initially occurs at the less sterically hindered para position. The mechanism is identical to before, nucleophilic addition followed by proton transfer. This is shown below:

The bromination of phenol occurs at room temperature without any Lewis acid to activate the electrophile. It starts with bromination of the para position through the standard two step electrophilic substitution mechanism with nucleophilic addition followed by proton transfer. But the reaction does not stop here …
A second and a third bromine add in the same fashion but at the two ortho positions. Again, the mechanism starts by feeding the oxygen's electrons into the ring to make it more nucleophilic.

Delocalization of the oxygen lone pair is such a good activating group that bromination does not stop after a single reaction but proceeds until three bromine atoms have added to the aromatic ring.
The hydroxyl group is said to be an electron-donating group or EDG and can be considered to be an activating group. If you draw the resonance structures of phenol you will see that the negative charge can be placed on three carbons. These carbons are electron rich and you can draw a resonance hybrid that has the partial charges on these carbon atoms. As these are the carbon atoms that are more electron rich they are more nucleophilic and the initial addition step occurs here.

The resonance structures of phenol show that there is an increased concentration of electrons on three carbon atoms. This means those three carbon atoms at more electron rich and hence are more nucleophilic. Reaction with an electrophile occurs here.
Electron donating activators, such as the hydroxyl group, are ortho,para directing groups as they promote reaction at the ortho and para positions.
Many students are confused by the concept of oxygen being an electron donating group. Lecturers spend a lot of time hammering home that atoms to the right of carbon are more electronegative than carbon and will polarize a σ bond by drawing electrons towards the electronegative atom (for example the oxygen atom of a carbonyl group pulls electrons away from the carbon making the carbon a good electrophile). Students then apply this concept to phenol and argue that oxygen is electron withdrawing and should deactivate the ring, turning it into a poor nucleophile. It is true, oxygen is pulling electrons out of the ring through the single/σ bonds. This is the inductive effect. But, as soon as there is a lone pair of electrons one bond away from a double bond (or π system) then the lone pair is in conjugation with the π system and delocalization is possible. Delocalization or resonance is invariably a stronger effect than the inductive effect. Delocalization is more important. Of course, there are always exceptions, and we'll see one of these later!
Other groups that add a conjugated lone pair of electrons will act as electron-donating or activating groups and will permit electrophilic aromatic substitution to occur. Aniline, the nitrogen equivalent of a phenol, is even more reactive, and the reaction occurs rapidly even at low temperature. Nitrogen is less electronegative than oxygen and delocalization of the lone pair of electrons is even easier.

Aniline is even more reactive than phenol, due to nitrogen being less electronegative. It is brominated three times even at low temperatures. It is virtually impossible to stop multiple bromination simply by changing the reaction conditions and it is necessary to change the structure of the molecule.
The mono-bromination of aniline is almost impossible. To overcome this limitation it is necessary to change the functional group. It is possible to temper or reduce the reactivity of aniline by forming an amide. The lone pair on nitrogen can now delocalize onto the carbonyl group. This results in less interaction with the aromatic ring as it is also being shared outside the ring. Now bromination occurs only once at the para position. It reacts para for steric reasons.

By converting aniline into an amide, its nucleophilicity is reduced. Now the nitrogen lone pair is also delocalized over the carbonyl group. This means it is less available to activate the aromatic ring and bromination only occurs once. The addition is in the para position for steric reasons.
Heteroatoms are not the only electron-donating groups, alkyl groups are also electron donating. Think about hyperconjugation and the stabilization of carbocations (if you haven’t covered this read the reactions of alkenes HERE). They are not as strong electron donating groups as heteroatoms as there is no delocalization of electrons. The weaker electron donation means the reactions require Lewis acids to proceed but the reactions are faster (about 1000 times faster) than that of benzene and alkyl groups are consider weak activating groups.

The bromination of toluene is faster than that of benzene. Alkyl groups are activating groups. They are ortho,para-directing.
Alkyl groups are ortho,para-directing groups. The directing effect cannot be explained by the delocalization of electrons and is normally justified by considering the stability of the cationic intermediate. With substitution in either the ortho or para positions there is a resonance structure that has the cation adjacent to the alkyl group. Tertiary cations are more stable than those that have the positive charge on a secondary carbon. Substitution at the meta position does not have the benefit of this stablization and is disfavored.

The methyl group of toluene is an o,p-directing group. The preference for attack in these two positions can be explained by the stability of the cationic intermediate and that attack at these two positions allows the cation to be drawn on the tertiary position.
When bromine adds to the meta position it becomes impossible to draw a resonance structure with the cation on the tertiary carbon. The intermediate is less stable than the other two, as it lacks the additional stability offered by a tertiary carbocation.
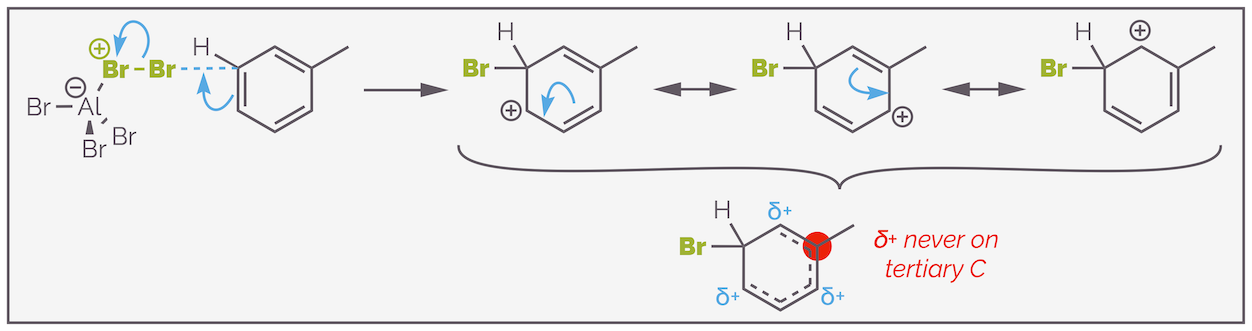
Bromination at the meta position lacks stabilization of the intermediate carbocation on the tertiary position. This means it is less favored (disfavored even).
You can either accept that alkyl groups are electron donating and weak activators that direct substitution to the ortho and para positions because of the stabilization of the cationic intermediate, or you can think about the frontier molecular orbitals and these will provide a nicer explanation (that was far too long a sentence).
Hyperconjugation or σ delocalization explains the activation of the benzene ring. As long as a C–H or C–C σ bond can align with the π bonds of the aromatic ring then the electrons can be thought to spread out or delocalize. It is not possible to draw resonance structures as you are dealing with σ electrons and this makes it a weak but still crucial effect. Unlike 'proper' resonance and delocalization, hyperconjugation doesn't exert an influence over a large distance. It activates the ortho positions more than the para, position simply because the ortho position is closer.

Hyperconjugation of the σ electrons activates the ring by making it more electron rich. The effect occurs over a short distance and the ortho position is more activated than the para position.
This same effect is responsible for the stabilization of the tertiary carbocation. As long as a C–H or C–C bond can align parallel with the empty 2p orbital of the cation then hyperconjugation or delocalization is possible. The positive charge is spread out over a larger volume and is more stable.

Alkyl groups can stabilize the cationic intermediate if a resonance structure places on the adjacent carbon (gives a tertiary carbocation). This can occur if substitution occurs in either the ortho or para positions. If the electrophile adds to the meta position then there are no resonance structures in which the empty 2p orbital can overlap with a σ bond of the alkyl group.
Electron donating groups activate the benzene ring, making it more nucleophilic and directing addition to the ortho and para positions. Electron withdrawing groups can pull electron density out of the ring making the ring less reactive. These are deactivating groups and they will direct addition to occur (if you force the reaction) at the meta position. An example is the bromination of nitrobenzene shown below:

The bromination of nitrobenzene occurs under forcing conditions (the reaction is heated over 100 °C), and bromination occurs at the meta position. Both the deactivation of the ring and the directing effect are due to the electron withdrawing nature of the nitro group.
The nitro group is an electron withdrawing group or EWG. It pulls electrons out of the ring and towards the electronegative oxygen atoms. The aromatic ring is less nucleophilic and less reactive. Electron withdrawing groups are said to be deactivating groups. The reactions of deactivated aromatic rings are slower and require more forceful conditions (stronger electrophiles or more vigorous reaction conditions).
It is possible to draw a series of resonance structures for nitrobenzene that place a positive charge on a ring carbon. You should see that the position of the positive charge alternates around the ring and it can be found in the ortho or para positions. This means the meta position is less deactivated or is more electron rich compared to the other two positions. As a result, electrophilic attack occurs at the meta position and electron withdrawing groups are meta directing.

The nitro group is electron withdrawing and deactivating. The resonance structures of nitrobenzene reveal that the positive charge can be distributed over three carbon atoms, the ortho and para positions. The resonance hybrid at the bottom highlights the most electron rich (meta) carbon atoms. These will act as nucleophiles.
Alternatively, electron withdrawing groups can be thought of as ortho and para avoiding groups. If you draw out the mechanism of electrophilic aromatic substitution for the reaction above three time, once for each of the different positions (good practice), then you will see that substitution at either the ortho or para positions places the cation of the cationic intermediate adjacent the electron withdrawing group. This is is highly disfavored, withdrawing electrons from an empty p orbital, a carbon with only six valence electrons is not going to happen! Only reaction at the meta position allows the cation to avoid being next to the destabilizing electron withdrawing group.

Electron-withdrawing groups can also be considered ortho,para-avoiding groups instead of meta-directing groups. If you inspect the various resonance structures that can be drawn for the cationic intermediate, it becomes clear that only the ones formed from reaction in the meta position avoid the disfavored interaction of having the cation adjacent to the electron withdrawing group (or in this example having two positive charges on adjacent carbon atoms).
So far things have been relatively straightforward, electron donating groups EDG are activating and they direct to ortho and para positions while electron withdrawing groups EWG are deactivating groups direct to the meta position. One set of functional groups misbehave and these are the halides. Fluorine, chlorine, bromine and iodine are deactivating groups but they are ortho and para directing. What is going on here? Simplistically, there is a competition between the inductive effect (the electronegativity of the deactivating group, or the idea that halides are electronegative) and delocalization of π electrons. The electronegativity of the halides deactivates the ring, withdrawing electrons and making it less nucleophilic. But, the halides have lone pairs of electrons and these can be delocalized into the ring making the ortho and para positions more electron rich. This latter effect is insufficient to activate the ring (you either argue poor orbital overlap or an inability of halides to support a positive charge) but sufficient to cause a directing effect.
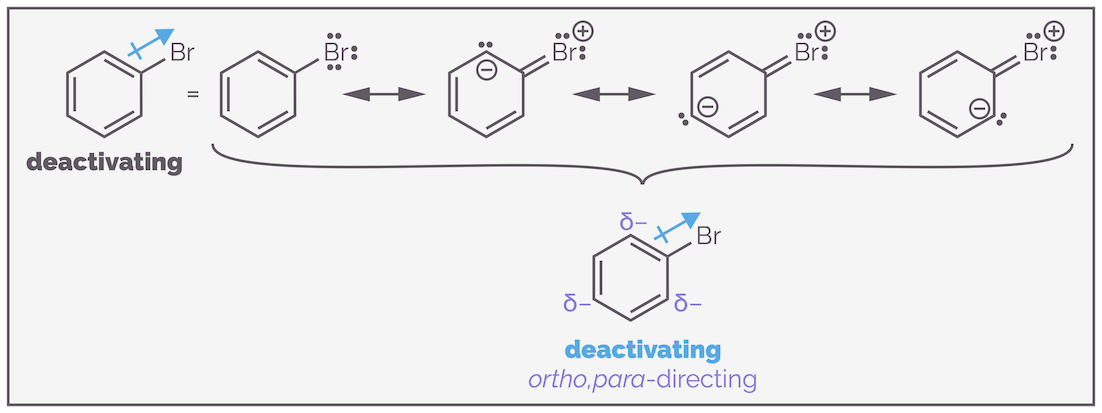
Halogens are the odd exception to our normal guidelines. They are electron-withdrawing groups that deactivate the ring by an inductive effect, but they are also ortho,para-directing through delocalization.
The various activating, deactivating and directing effects are summarized in the chart below:
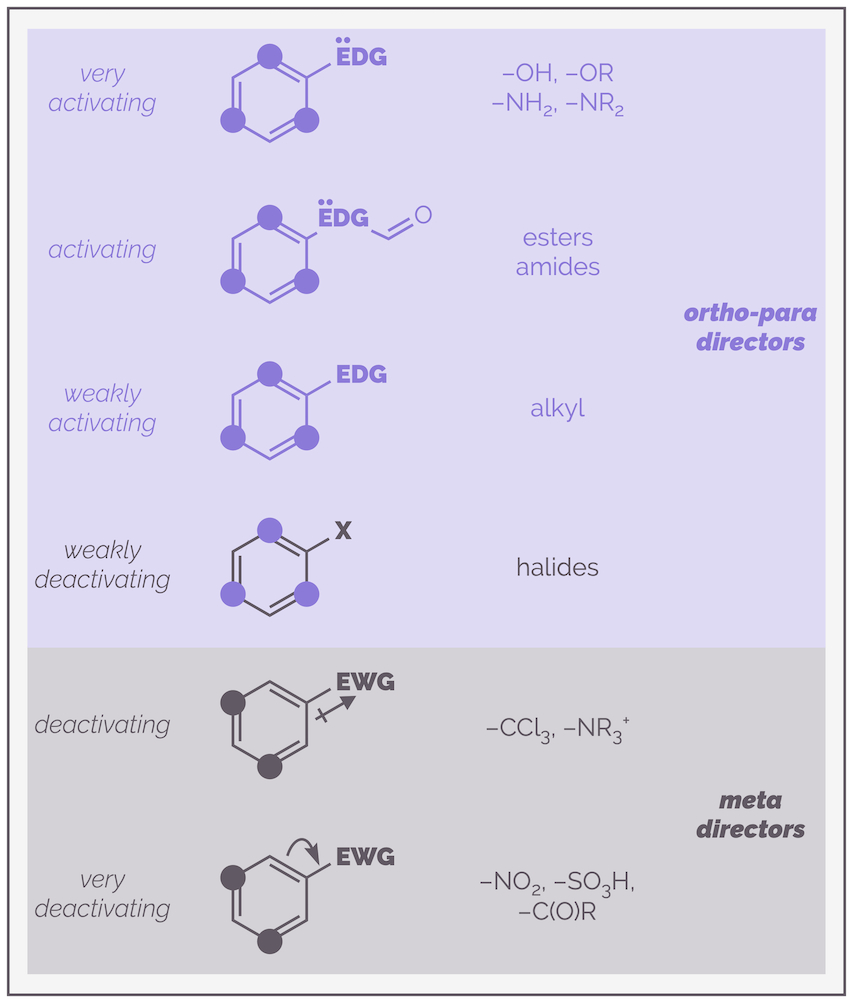
A chart summarizing the various effects of adding substituents to a benzene ring.
Electrophilic Aromatic Substitution is a General Reaction
Above, you worked through the mechanism for the bromination of benzene. The same mechanism holds true for any strong electrophile.

Electrophilic aromatic substitution is a general reaction. There are invariably three steps. The first (step 0) is activation of the electrophile, this is necessary for all but the most activated benzene derivatives. Step 1 (of the actual substitution reaction) is nucleophilic addition in which the aromatic ring attacks the electrophile. This breaks the aromaticity of the ring and creates a cation intermediate. Step 2 is deprotonation or proton transfer and this regenerates the aromatic ring.
The only really difference in all the examples you will see below is Step 0, the activation of the electrophile, otherwise the reactions are identical. Activation can be achieved using a strong acid (in a dehydration) or with a Lewis acid, and invariably leads to the formation of a cationic species. A powerful electrophile (or activated nucleophile) is required for the subsequent steps. After that, the mechanism of electrophilic aromatic substitution (SEAr) is the same for all the reactions. Step 1 is nucleophilic addition to give the cationic intermediate followed by Step 2, a proton transfer and re-aromatization of the ring.
The most common examples are given below:
Nitration
Sulfonation
Friedel-Crafts alkylation
Friedel-Crafts acylation
Nitration
Nitration is the addition of a nitro (NO2) group to the aromatic ring. It is normally achieved with a mixture of nitric acid (HNO3) and a strong acid such as sulfuric acid (H2SO4).

Nitrobenzene can be formed by nitration of benzene with a mixture of nitric and sulfuric acids.
The strong acid must be more acidic than nitric acid as it has to donate a proton to nitric acid to create a good leaving group. The protonated nitric acid expels water in a dehydration reaction, and this leads to a cationic, or reactive, electrophile called a nitronium ion (NO2+).

Nitric acid is activated by protonation with a stronger acid, most commonly sulfuric acid. A dehydration process occurs to give a nitronium cation, the activated electrophile.
Once the activated electrophile is formed, the standard reaction mechanism occurs. The nucleophilic benzene ring attacks the nitronium species to break the aromaticity and give the cationic intermediate. Deprotonation reforms the aromatic ring and gives the nitro compound.
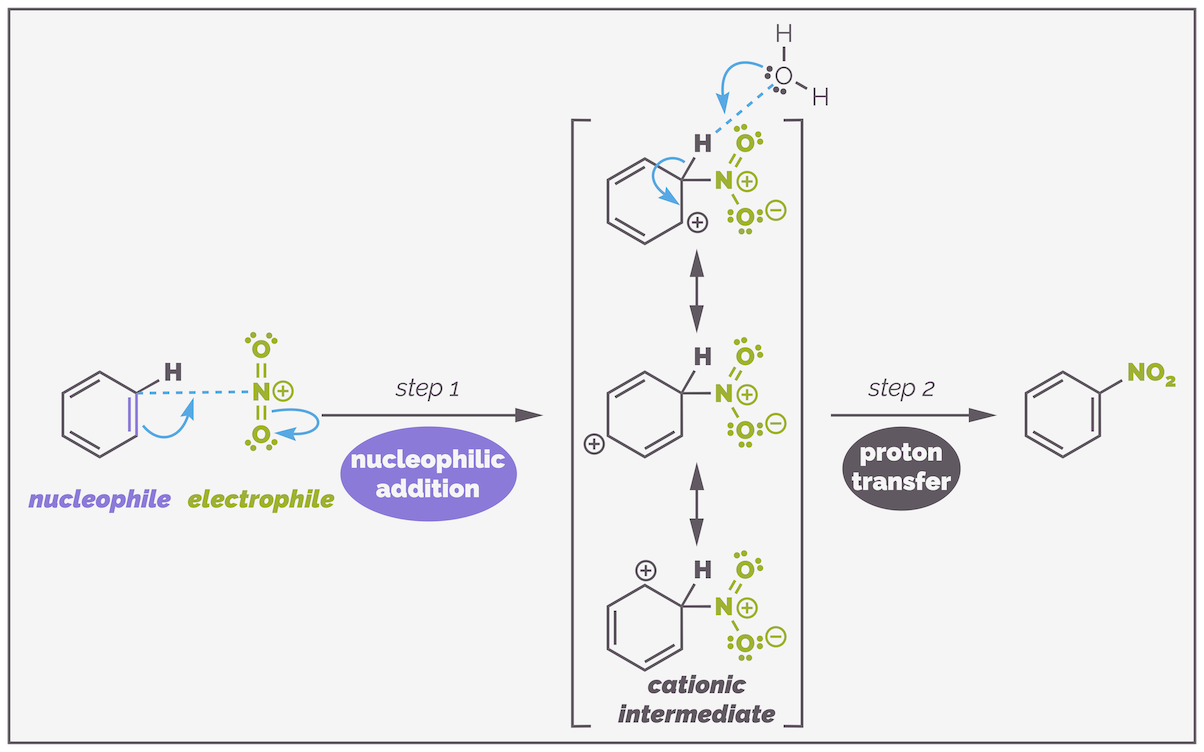
The nitration mechanism is another standard example of electrophilic aromatic substitution. The electrophile is activated in a prior step, this is followed by nucleophilic addition with the aromatic ring attacking the nitronium ion. The cationic intermediate is deprotonated in the second step to regenerate the aromatic ring and give the product.
Nitration is a reliable means of introducing a nitrogen atom onto a benzene ring, but to be useful, you need a method of converting the nitro group into an amine. This is achieved by nitro reduction. The two most common methods are using a metal in a dilute acid (often tin in HCl but changing the metal (iron or zinc) allows tuning of the redox potential and can lead to improved selectivities in the presence of groups that can be reduced, such as halides) or hydrogenation with a palladium catalyst supported on charcoal (H2, Pd/C).

The nitro group can be reduced to an amine and this provides a useful way to introduce the nitrogen atom onto an aromatic ring. It also offers opportunities to alter directing effects.
Amines are activating groups, directing to the ortho and para positions while the nitro group is deactivating and directs to the meta position. The ability to change from one to the other gives a chemist a great deal of control in adding subsequent substituents.
Sulfonation
The sulfonation of an aromatic ring is very similar to nitration and allows the addition of a sulfonic acid group (R–SO3H). This can be achieved by reacting benzene with fuming sulfuric acid or sulfur trioxide and sulfuric acid.

The sulfonation of benzene to synthesize benzenesulfonic acid.
Sulfur trioxide is a powerful electrophile and it is probable that the sulfonation does not require activation of the electrophile prior to electrophilic aromatic substitution. In such a case, the final step of the mechanism would be protonation of the sulfonate ion (ArSO3–). Under acidic conditions, proton transfer isn't an issue. Instead though, I have drawn a mechanism that involves protonation first. This is simply to draw the connection with the previous reactions; activation of the electrophile is followed by electrophilic aromatic substitution.

One of the possible mechanisms for sulfonation of benzene involves protonation of sulfur trioxide to create the activated electrophile. This participates in the standard electrophilic aromatic substitution mechanism. Arguably, sulfur trioxide is already sufficiently activated to take part in this reaction without prior activation. To draw this mechanism simply add step 0 the proton transfer to the end of the reaction sequence and protonate the sulfonate anion formed by nucleophilic attack on sulfur trioxide.
Sulfonic acids are useful functional groups. They are strong acids, with toluenesulfonic acid having a pKa of –2.8 in water (considerably stronger an acid than benzoic acid pKa = 4.2). Salts of sulfonic acids, sulfonate anions, are frequently used to make organic compounds more water soluble or create detergents.
Another useful property of sulfonic acids is that their introduction onto an aromatic ring is reversible. If you use dilute sulfuric acid instead of the fuming (concentrated) sulfuric acid and heat, then the reaction can be reversed. This becomes useful if you need to temporarily block reaction at certain positions on a ring.

The sulfonation of aromatic rings is an equilibrium process, that is the reaction can go both forwards (addition of the sulfonic acid) and in reverse (removal of the sulfonic acid). The position of the equilibrium can be controlled by the reaction conditions. Fuming sulfuric acid leads to sulfonation. Use of dilute sulfuric acid leads to removal of the sulfonic acid.
Friedel-Crafts Alkylation
This substitution reaction can be used to form C–C bonds. Again, the key to the reaction is creating a sufficiently active electrophile, in this case, a reagent that behaves as if it was a carbocation (and in many cases probably is a cation). The Friedel-Crafts alkylation is essentially the same as bromination, a Lewis acid, normally aluminium trichloride is used to activate an alkyl chloride. The activated electrophile is then attacked by the aromatic ring in the standard electrophilic aromatic substitution reaction.

The Friedel-Crafts alkylation is the reaction of an alkyl chloride with an aromatic ring to give a new C–C bond.
The activation step is the same as bromination with a lone pair of electrons from the halide attacking the Lewis acid. It is possible that the tertiary carbocation (or other stabilized cations) shown below is formed but for primary carbocations it is almost certainly the aluminium complex that is the actual electrophile.

First the electrophile must be activated (Step 0). This is achieved by the formation of a carbocation (or complex that behaves as a carbocation).
The carbocation is sufficiently electrophilic to be attacked by the aromatic ring in the nucleophilic addition step (Step 1). Finally, deprotonation re-forms the aromatic ring and gives you the product. Any base will achieve the deprotonation as the driving force is re-aromatization. I have shown the chloride anion formed during the activation (probably should have shown the aluminate complex).
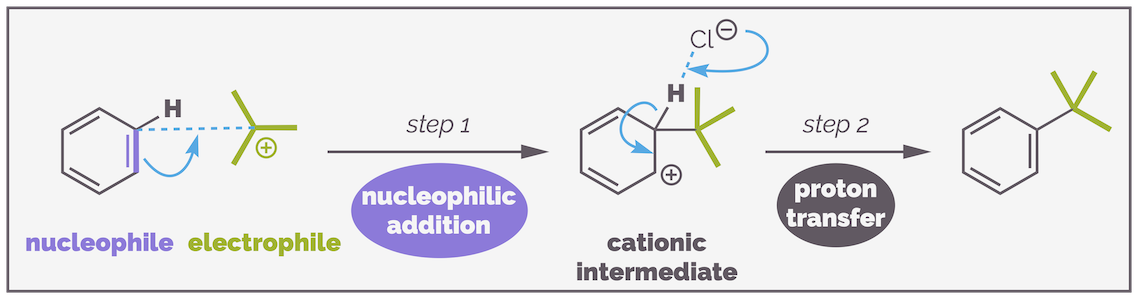
The Friedel-Crafts alkylation is another example of electrophilic aromatic substitution. The mechanism is identical to all the previous examples.
There are two problems with the Friedel-Crafts alkylation. The first is that it is difficult to control the number of additions that occur. Alkylation of a benzene ring adds an electron-donating group and makes the ring more nucleophilic or reactive. The product of the initial alkylation will frequently react a second time (and potentially more).

The Friedel-Crafts alkylation adds an electron-donating alkyl group. The inductive effect (or hyperconjugation) feeds electrons onto the ring making it more nucleophilic and making the second addition faster.
The second problem is that the Friedel-Crafts alkylation only works well with stable cations such as the tertiary carbocation drawn above. Primary carbocations are not stable and are hard to form. The reaction with 1-chloropropane results in a mixture of compounds formed by rearrangement and multiple additions.

The Friedel-Crafts reaction of primary alkyl halides is notoriously problematic due to the instability of primary carbocations.
It is unlikely that the primary carbocation ever forms. Instead, the minor product is prepared by a substitution reaction in which the nucleophilic aromatic ring directly attacks the Lewis acid complex.

Primary cations are hard to form and the Friedel-Crafts alkylation is more likely to occur through direct displacement of the appropriate Lewis acid adduct. I have called step 1 a nucleophilic addition so that it looks the same as all the other reactions (it is the same), but the reaction is technically a substitution as the aromatic ring replaces the metal complex … but this then confuses people as the reaction is still an addition-elimination (deprotonation) process.
While a primary carbocation will not form, there is a way to create a more stable secondary carbocation and this leads to the major product. Instead of the aromatic ring attacking the Lewis acid adduct and hydride (or H–) shifts, displacing AlCl4– and leaving behind the secondary cation. This reacts in electrophilic aromatic substitution.

The precursors to primary carbocations can rearrange to give secondary (or tertiary) cations. This will happen prior to electrophilic aromatic substitution and leads to the major product of reaction.
Friedel-Crafts Acylation
The solution to both problems, multiple additions and rearrangements, is the Friedel-Crafts acylation reaction. This reaction has numerous advantages over the Friedel-Crafts alkylation and allows you to add primary alkyl groups to aromatic rings (and so much more). The Friedel-Crafts acylation uses an acyl chloride instead of alkyl chloride.

The Friedel-Crafts acylation gives aromatic ketones (and carboxylic acid derivatives).
The Friedel-Crafts acylation solves the issue of multiple additions by adding an electron withdrawing group to the aromatic ring. The ketone carbonyl group is in conjugation with the aromatic ring allowing the electrons to be delocalized. This deactivates the ring and only a single addition occurs.
The rearrangement is prevented as the activated electrophile, the cation, is stabilized by delocalization across the carbonyl group. The acylium ion is sufficiently stable that there is no driving force for rearrangement.

The mechanism for Friedel-Crafts acylation follows the same pattern as before. There is activation of the electrophile followed by nucleophilic attack and proton transfer. The advantage of the Friedel-Crafts acylation is that the acylium ion is stabilized so there is no rearrangement and the addition of a carbonyl group to the aromatic ring deactivates the ring to subsequent reactions.
The product of the Friedel-Crafts acylation can be transformed into a simple alkane by reduction of the ketone. The Clemmensen reduction uses zinc amalgam (written as Zn(Hg)) in concentrated hydrochloric acid (HCl) to completely remove the carbonyl oxygen. The mechanism of the reduction is beyond undergraduate studies (and I don't believe that it is definitively known). But the sequence of Friedel-Crafts acylation followed by reduction is a reliable method for forming the product of the Friedel-Crafts alkylation of primary halides, a reaction that cannot normally be achieved.

The Clemmensen reduction converts ketones into alkanes.
Multiple directing groups and the synthesis of aromatic compounds
So far, you have looked at the common reactions of aromatic rings and seen how activation of various electrophiles leads to electrophilic aromatic substitution. You should also be able to divide substituents on an aromatic ring into those that activate the ring and are ortho, para-directing, or those that are deactivating amd meta directing. The last two subjects I want to discuss are, what happens if there are multiple directing groups on a ring and how do you make the aromatic derivative that you want?
When there are multiple substituents on a ring there are two possible situations; the substituents are either complementary, directing reaction to the same position, or they work against each other or are competitive. The first situation is easy to deal with as the directing effects reinforce each other. For example, bromination of the compound below occurs ortho to the phenol. The phenol is activating and directs to the ortho and para positions but the latter is blocked by the aldehyde. The aldehyde is deactivating and directs to the position meta to the aldehyde, which is the same position and is ortho relative to the phenol. They both cause reaction at the same place.

Two substituents can direct reaction to occur at the same position. Such groups are complementary. Note now the name of the positions around the ring changes depending on the which group we are referring to. The ortho,para position is relative to the phenol hydroxyl group while the meta positions is relative to the aldehyde that causes this directing effect.
The alternative is that the groups can work against each other and direct to different positions as in the example below:

Both groups activate the ring to electrophilic aromatic substitution and both are ortho,para-directing relative to their own positions. The phenol directs the reaction as it is a stronger activating group, and bromination is ortho to the hydroxyl group and meta to the alkyl group.
Both the phenol and the alkyl group are electron donating so both activate the ring and direct ortho,para to themselves. The phenol is the stronger activating group as delocalization of π electrons is a more important effect than induction or hyperconjugation. The stronger activating group directs the position of attack as it makes these carbons more electron rich.
Strong activating groups dominate selectivity. If a molecule contains both an activating group and a deactivating group, it is the activating group that controls the position of reaction. It promotes the reaction by feeding electron density into the ring and on to specific carbons (as shown by drawing the resonance structures), the electrophile will react on these electron rich carbons.

The strongest activating group, the one that donates more electron density into the ring will control the reaction. It controls the reaction as it encourages substitution. In the example above the phenol directs reaction to occur at either the ortho or para positions. The ortho position wins due to steric hindrance.
The phenol group controls the reactivity of the example above. It is activating the ring towards electrophilic aromatic substitution and this is more important than the deactivating effect of the nitro group. As a result, the bromine is added to either the ortho or para positions. The position ortho to both the hydroxyl group and the nitro group is too sterically demanding for reaction to occur. The position ortho the hydroxyl group is probably more electron rich (further from the nitro group) and less sterically demanding so that under standard conditions (bromine in acetic acid) reaction occurs predominantly at this position.
Controlling regioselectivity
There are a number of tricks that allow chemists to control which position is functionalized during electrophilic aromatic substitution reactions. The first of these is the use of sulfonic acids as blocking group. The major product of the nitration of anisole is the para-derivative.

The nitration of anisole occurs predominantly at the para position due to steric hindrance slowing addition at the ortho position.
But, what if you wanted the ortho-isomer, how might you make this? One route relies on the reversibility of the sulfonation reaction. Using concentrated sulfuric acid results in para sulfonation. Nitration of the resulting compound has to go ortho to the ether. In this example, both the functional groups direct to the same position. The ether is activating an ortho,para directing while the electron withdrawing sulfonic acid is deactivating and meta directing. Finally, desulfonation can be achieved by heating the compound in dilute sulfuric acid.

Sulfonic acids can be used as a blocking group that prevents reaction at specific positions but can be removed at a later stage.
The second trick uses exploits your ability to change certain electron withdrawing groups into electron donating groups to control selectivity. The two regioisomers below can be selectively synthesized by selecting either the amine or the nitro group as the starting material.

How could you selectively make each of these regioisomers.
If you want para-chloroaniline you would start from aniline. The first step would be amide formation. This tempers the reactivity, making the aniline less activated and permitting a single chlorine atom to be introduced at the para position. The para position is favored over the ortho position to avoid steric interactions. Hydrolysis gives the desired product.

The para-isomer is synthesized by first forming the amide. This prevents over chlorination and promotes chlorination at the para position as the amide is weakly activating, ortho,para-directing but the bulk of the acetamide hinders approach to the ortho positions.
The other regioisomer starts from nitrobenzene. The electron withdrawing nitro group directs reaction to occur at the meta position. Following electrophilic aromatic substitution, the nitro group is reduced to give the desired product.

The synthesis of the meta-regioisomer starts from nitrobenzene. The electron withdrawing nitro group is meta directing, so delivers the chlorine to the correct carbon atom. Selective reduction of the nitro group (annoyingly, aryl halides can be reduced with metals in weak acids, so you must choose the correct combination, which can be determined from standard electrode potentials (something I hope I never have to discuss on here!)) gives the desired isomer.
Similarly, the product of Friedel-Crafts acylation is a ketone, which is deactivating and meta directing. Chlorination can be followed by Clemmensen reduction to give the meta-isomer. Alternatively, you can reduce the ketone to an alkyl group first. Now you are chlorinating a weakly activating group. This is is ortho,para-directing. Depending on the order of reactions you can synthesize either isomer.
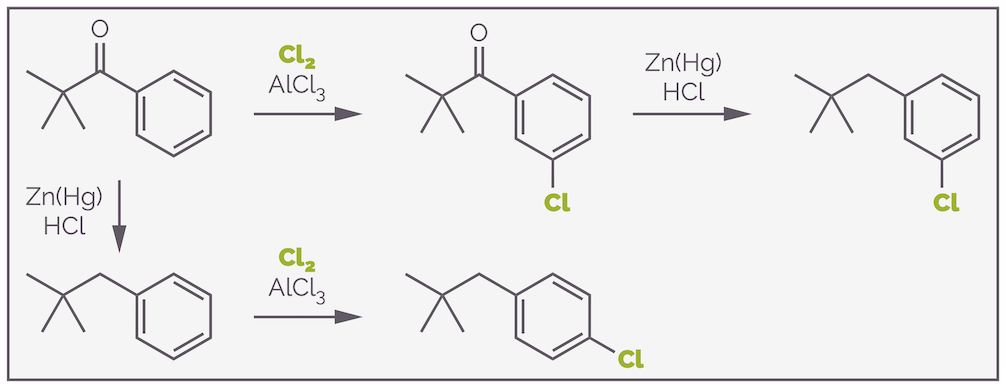
Switching between a ketone and an alkyl group allows control of regiochemistry.
Conclusion
Electrophilic aromatic substitution is a general reaction that exchanges a hydrogen atom on an aromatic ring with a powerful electrophile. The most common examples allow the formation of C–X, C–N, C–S and C–C bonds. The most common examples are summarized in the scheme below:
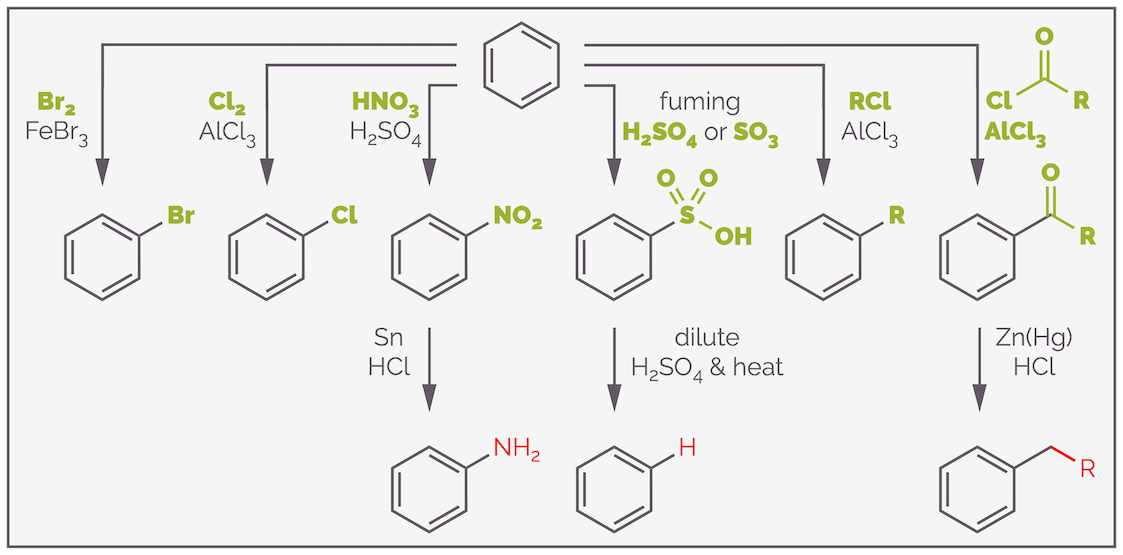
The common examples of electrophilic aromatic substitution and useful functional group interconversions of the resulting products.
Different substituents on the benzene ring can alter the reactivity dramatically. Simplistically, substituents can be divided into two categories, groups which activate the ring and those that deactivate it. Activating groups are electron rich and they increase the nucleophilicity of the ring, making reactions faster. All activating groups direct substitution to the ortho and para positions. Most deactivating groups are meta directing. The halides are the exception. These are deactivating but are still ortho and para directing.

A summary of activating and deactivating groups along with the position they direct reaction to.
If there are multiple substituents on a ring the activating group controls the reaction. Substituents that can delocalize a lone pair of electrons into the ring are the strongest activators and have the greatest influence on directing reactions.
Electrophilic aromatic substitution is an important reaction for the formation of molecules.