An Introduction to Enols & Enolates
Introduction
The second reaction I covered for this blog, after the proton transfer of acid/base reactions, was nucleophilic addition to the carbonyl group. The polarity of the C=O double bond, along with the relative weakness of the π bond makes this group an excellent electrophile. I think a lot of organic chemistry principles can be covered when discussing carbonyl-containing compounds as electrophiles.
The carbonyl group is often an excellent electrophile due to the polarization of the bond and the ease of breaking the double bond.
But carbonyl-containing compounds can also behave as nucleophiles. The polarity of the bond, amongst other factors, permits the carbonyl group to activate the adjacent carbon atom and turn it into a nucleophile (and the oxygen atom can also behave as a nucleophile). The nucleophilic form of the carbonyl group is the enol, along with a charged, more activated, form known as the enolate.

The carbonyl group can make a good nucleophile. Under neutral or acidic conditions, it can exist in the enol form, which is nucleophilic through the adjacent carbon atom. Under basic conditions, it is converted to an enolate, a more powerful nucleophile.
I need to stress that this is just a simple introduction to this key chemistry. The reactions of enols and enolates are one of the cornerstones of organic synthesis. Undoubtedly, there will be more summaries cover more of this chemistry in considerably more detail. This is just the start. The examples in this summary will be simple. I will only be looking at simple reactions of the enols/enolates at the carbon atom, I will not even show a reaction at the oxygen atom. I will not be discussing stereochemistry (except to mention that enols and enolates cause racemization at the adjacent carbon atom). I will not be discussing enol/enolate equivalents, such as enamines, and I will not be talking about the aldol reaction (and all its variants) even though it is one of the most useful reactions out there. All these topics will get their own summaries (which is something I look forward to writing, and for once that was not my sarcastic typing).
So without any more pre-amble let’s introduce this topic …
Enols
Aldehydes and ketones that possess a C–H bond on the carbon adjacent to the carbonyl group can exist in equilibrium with a structural isomer known as an enol. The name comes is derived from the C=C double bond or alkene (-ene) having a hydroxyl or alcohol (-ol) group attached to it, but don't think of it as an alkene and an alcohol, it is its own functional group with its own reactivity (see the rest of this summary!).

Keto-enol tautomerization describes the equilibrium that exists between functional groups containing a carbonyl group and the enol form where a proton has shifted. The two compounds are structural isomers, not resonance structures. The equilibrium normally favors the carbonyl compound. In the case of acetone, there is only a trace of the enol tautomer, but its existence is still very important.
The two molecules are structural isomers that differ by the position of a single proton and a double bond. Such isomers are known as tautomers and the interconversion of the two is called tautomerization. Tautomerization is a general process that does not require a carbonyl group (you can see it when a proton transfers from one nitrogen to the other of imidazole). In this example, as there is a carbonyl and an enol in equilibrium, the reaction can also be (more specifically) called enolization or keto-enol tautomerization.
The two structures are related by an equilibrium arrow. This is not an example of delocalization and these are not resonance structures. A chemical reaction is occurring and the molecules are distinct structural isomers. For simple aldehydes and ketones the equilibrium favors the keto form or the carbonyl group. This is easy to explain if you think about the bonds in the two isomers. While an O–H bond is slightly stronger than a C–H bond (440 kJ mol–1) versus 463 kJ mol–1, a carbonyl bond (C=O) is much stronger than an alkene (C=C; 720 kJ mol–1 versus 620 kJ mol–1) due to the polarization of the carbonyl group. The carbonyl containing isomer is more stable and predominates.
The situation is often reversed when the enol form is stabilized. In the example below, the enol is in conjugation with the phenyl ring, and there is additional stabilization by a very favorable intramolecular hydrogen bond. Now the enol form is favored.
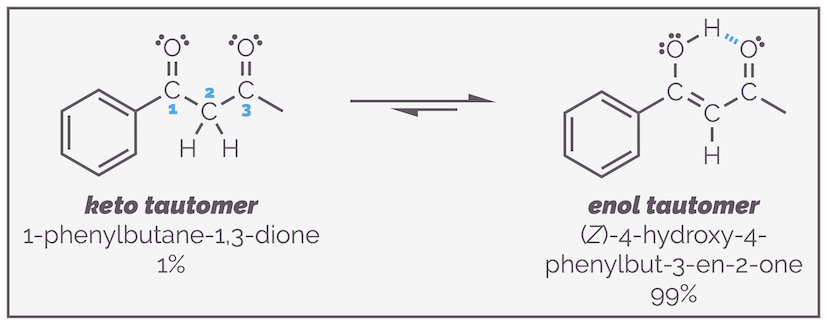
In the case of a 1,3-dicarbonyl compound (so named as the carbonyl groups are on the first and third carbons in a row, as shown by the blue numbering), the enol form of one of the carbonyl groups is favored over the keto form. The alkene is stabilized by conjugation with both the benzene ring and the second ketone. The enol is also stabilized by the internal hydrogen bonding.
The combination of conjugation and internal hydrogen bonding causes 1,3-dicarbonyl (or β-dicarbonyl) compounds to favor the enol tautomer. Again, with the example below, I want to stress that the three tautomers are three separate molecules that differ by the movement of a hydrogen atom. They are connected by equilibrium arrows as they are the result of a reaction. I have also drawn delocalization of one of the structures to show that the two resonance structures differ by the position of the double bonds and formal charges but not the movement of atoms.

A second example of a 1,3-dicarbonyl compound in which the enol form is more stable than the keto form. This diagram emphasizes that tautomerization is the movement of a proton and π bonds while delocalization is only the movement of electrons. There are three separate structural isomers shown along with one resonance structure of the last isomer.
The middle tautomer is the favored compound, with 76% of the mixture existing in the form. It is more stable than the others as the alkene is more substituted (see HERE), while the internal hydrogen bonding and conjugation favor the enol over the dicarbonyl. In this example, if you were to record the 1H NMR spectrum of the mixture you would observe two sets of peaks, one set for each of the enol tautomers (the sensitivity of common NMR spectrometers found in universities is such that you normally require around 5% of a compound before you observe it).
Before I go any further, I need to introduce the common naming system when discussing carbon atoms or positions relative to a carbonyl group. The carbon adjacent to the carbonyl group is know as the α-carbon (alpha-carbon). The hydrogens on this carbon atom are the α-hydrogens. The second carbon is the β-(beta)-carbon and the third is the γ (gamma) carbon. Another name for a 1,3-dicarbonyl compound, such as the one in the example above, would be a β-keto aldehyde as the ketone is on the second or β-carbon.

Just to make things simple, chemists like to name the carbon atoms next to a carbonyl group using the Greek alphabet. This means the first carbon adjacent to the carbonyl is α and then it keeps going through β, γ, and δ. Chemists also use numbers (as attested by having 1,3-dicarbonyls as well as β-keto carbonyls).
If you mix a simple ketone, such as acetone with deuterated water (D2O) and record the 1H NMR over time, you will observe that the peaks change. Very slowly the hydrogen atoms are replaced by deuterium atoms. This reveals that the keto-enol tautomerization occurs even if you can’t see the enol isomer. This observation can be explained by looking at a mechanism for tautomerization:

There is a slow exchange of deuterium and hydrogen from D2O and an enolizable carbonyl group. This leads a change in the 1H NMR spectrum over time.
This process and the mechanism is important for two reasons; the exchange of deuterium and hydrogen is evidence that keto-enol tautomerization occurs. Secondly, it shows that the process is intermolecular. Often, you will see the process drawn as an intramolecular process, as shown below, this is incorrect. The atom (and more importantly, the orbitals) are almost certainly too far apart to permit such a transfer.
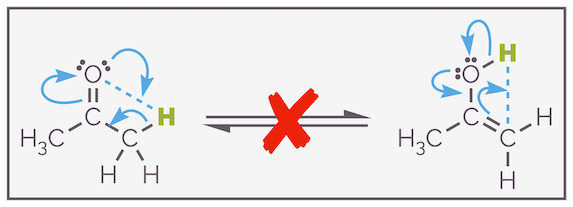
Keto-enol tautomerization is not an intramolecular process even though this is easier (and lazier) to draw.
The deuterium-hydrogen exchange shown above is a slow process, and noticeable changes in spectroscopy only occur after an appreciable amount of time. The reaction can be accelerated by either acid or base catalysis. The acid catalyzed reaction is similar to the mechanism above except that the hydronium ion acts as the proton source and water as the base:

Acids will catalyse keto-enol tautomerization through a cation intermediate. The reaction occurs through a protonation and then deprotonation step. The cationic intermediate is delocalized and best represented by two resonance structures.
The mechanism involves two proton transfers. In the first step the acid protonates the carbonyl group. Acid catalysis results in the formation of a cationic intermediate. The electrons of this intermediate are delocalized and you can draw two resonance structures. This is not a step of the reaction. Remember, resonance structures just give a more accurate representation of the intermediate. Either resonance structure can be used in the second step, which is another proton transfer and this time it is a deprotonation to give the enol.
The base-catalyzed reaction is very similar. Again, it occurs through two proton transfers. Again, the intermediate is best represented by two resonance structures. There are two differences, the intermediate is negatively charged or is an anion, and the reaction occurs by a deprotonation and then a protonation (so the proton transfer steps are reversed). The anionic intermediate, the enolate, is very important. You will be using this in many reactions in the rest of this summary and subsequent summaries.

Keto-enol tautomerization can also be base catalyzed. Deprotonation, to give the important enolate anion, occurs first, and is then followed by protonation to give the enol. The anionic intermediate, the enolate, is delocalized and best represented by two resonance structures.
Like all academics (techically, I’m an ex-academic), I have a list of common mistakes. One that always crops up is using both a hydroxide anion and a hydronium cation in the same reaction. This isn’t going to occur, they would react with each other rather than the substrate (not strictly true if you have been through the acid-base summary but the equilibrium is far to one side!). The key is to to remember that the acid catalyzed process always goes through neutral or cationic species and the base-catalysed reaction always has either neutral or anionic species.
Reactions tend to use conjugate acid-base pairs. If the reaction is acid catalyzed use the hydronium ion, H3O+ (the acid), to protonate the substrate and water, H2O (the conjugate base), to deprotonate the substrate. With the base-mediate process, use hydroxide, HO– (the base), to deprotonate and water, H2O (the conjugate acid), to protonate.
Before looking at the reactions of enols and enolates, let’s summarize which carbonyl-containing molecules can form nucleophiles. Enols or enolates are only possible if there is an α-hydrogen atom. No α-proton, no nucleophile. I know this sounds obvious but I’ve seen far too many mistakes to not want to emphasize this. Below are some examples of compounds that can or can’t enolize. Make sure you can identify the key proton.
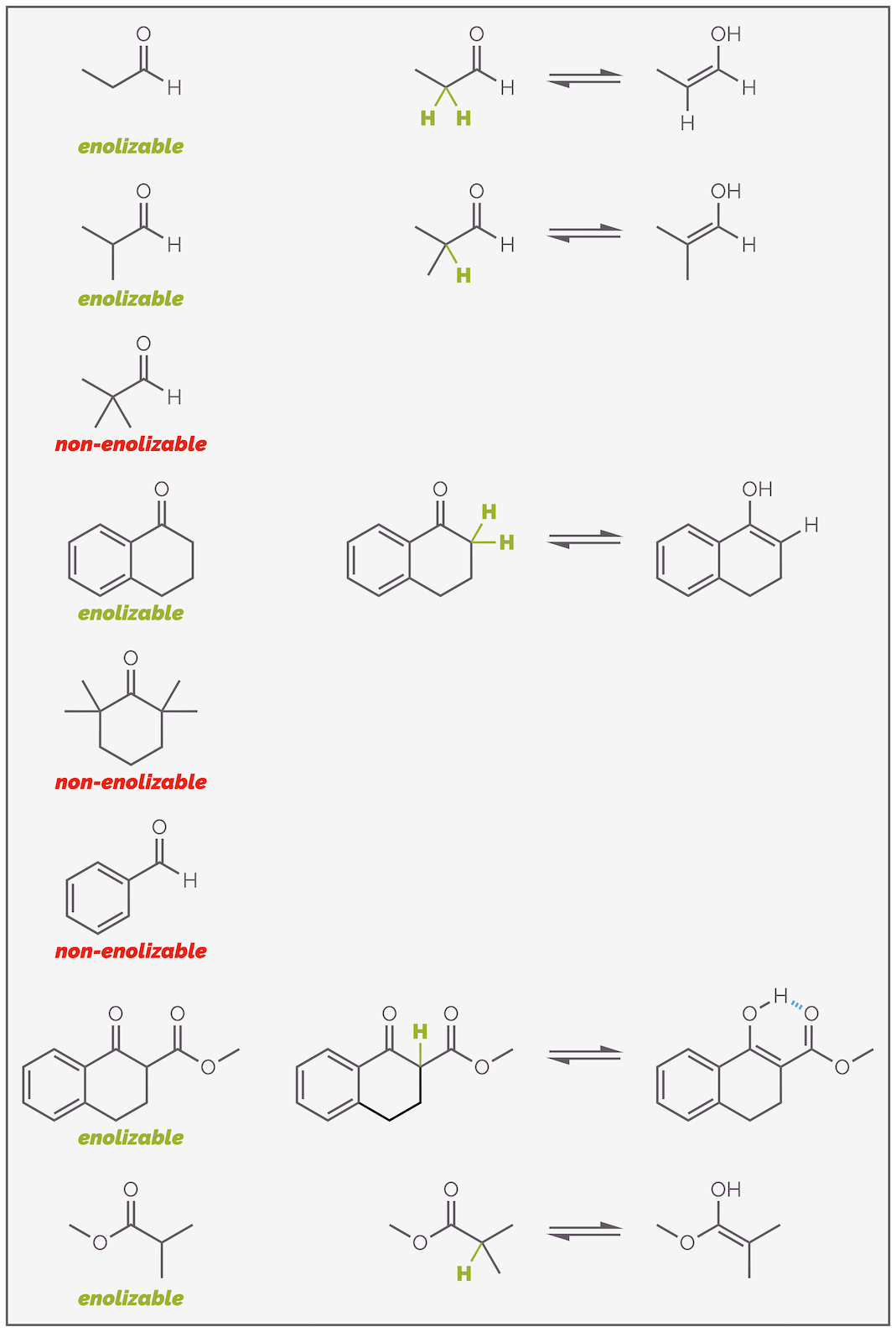
Here are a number of examples of carbonyl-containing compounds. Only those with an α (alpha)-hydrogen are enolizable.
The Reaction of Enols
1. Protonation (racemization)
Enols are nucleophiles. The diagram below shows the enolization of acetone to given an enol. The electrons of an enol can delocalize and the structure is best represented as two resonance structures. One of these shows a negative charge on the α-carbon. This shows that the carbon is electron rich and will behave as the nucleophile. Most reactions occur so that the new bond forms to the carbon but there are also two lone pairs of electrons on the oxygen that can act as a nucleophile too. This is rare, oxygen is more electronegative and one lone pair is delocalized, and there will be no examples of these reactions in this summary (see what I did there? We will have to discuss these reactions at some point).

Enolization of acetone gives the nucleophilic enol. The resonance structures of the enol show that that carbon atom has partial negative charge and is nucleophilic.
The simplest reaction of an enol is protonation or the reverse of enolization, converting the enol to the carbonyl. At first glance, this might not seem an important reaction (and you have already covered it above), but it can have significant consequences if the α-carbon is a stereocenter (see Summary HERE). If the initial substrate is enantiomerically enriched, meaning one enantiomer predominates then enolization followed by protonation can lead to racemization and the formation of a racemic mixture of both enantiomers. You have lost stereochemical purity.

Enolization of a carbonyl-containing compound with an α-stereocenter can lead to racemization or epimerization. The mechanism for acid-catalysed enolization is shown and the final proton transfer can occur from either face of the enol. This leads to the formation of a mixture of enantiomers.

The enol is flat with the carbon atoms being trigonal planar. Protonation can occur from either face (top or bottom in this diagram) and this leads to the different enantiomers.
Both the acid- and base-catalyzed reactions take a tetrahedral α-position and converted it to a trigonal planar flat carbon with no stereochemical information. The protonation of the enol can occur from either face of the double bond and this leads to the formation of a mixture of enantiomers or racemization. If the substrate contains multiple stereocenters then the reaction will only change one of them, and you are not forming a new enantiomer but a diastereomer. Now the reaction is known as epimerization.
The mechanism for the base-catalyzed process is the very similar, the only difference is that the nucleophile that is protonated is an anion.

Enolization of a carbonyl-containing compound with an α-stereocenter can lead to racemization or epimerization. The mechanism for base-catalyzed enolization is shown and the final proton transfer can occur from either face of the enol. This leads to the formation of a mixture of enantiomers. The process is almost identical to that of acid-catalyzed racemization.
2. Bromination (halogenation)
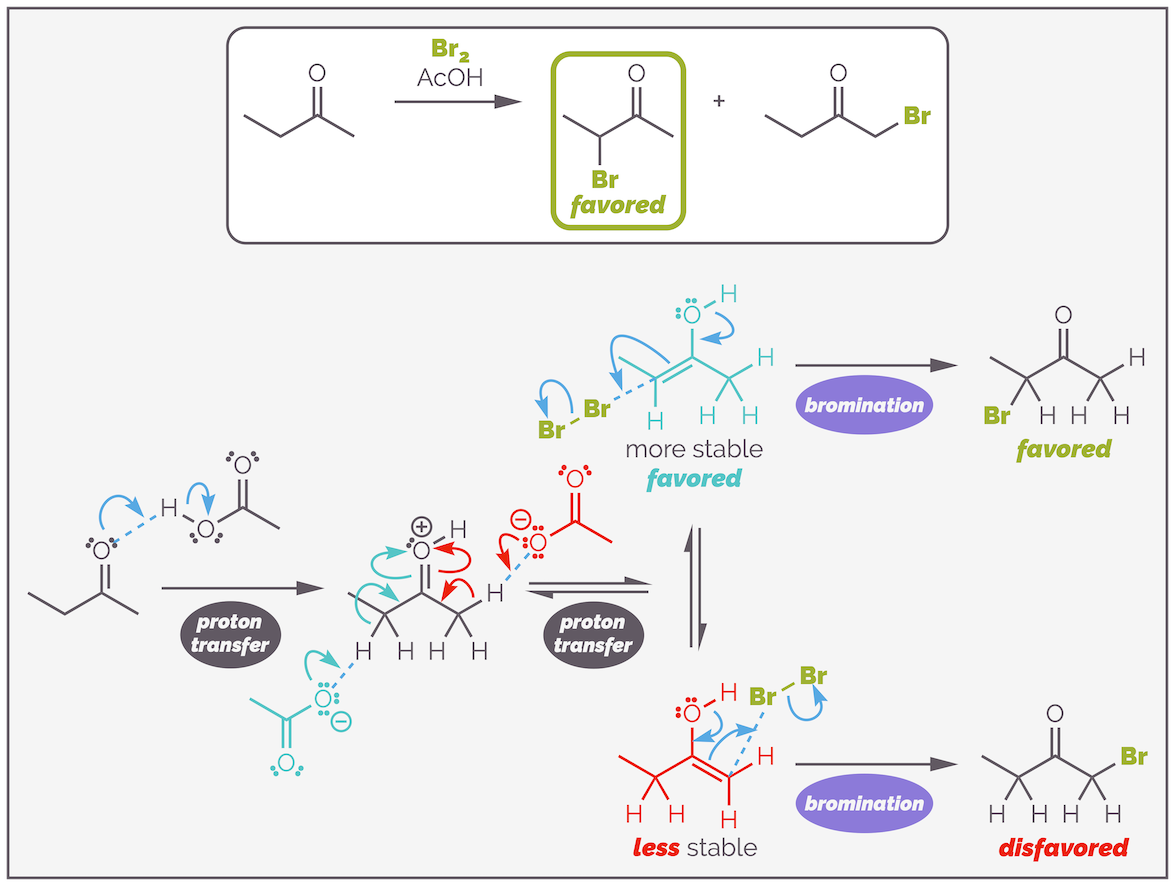
A common example of a reaction of an enol that leads to a new product is bromination (actually, this should be halogenation as the reaction works for chlorine and iodine as well). In this reaction, the small amount of nucleophilic enol formed in the standard equilibrium, reacts with the electrophilic bromine and this leads to the α-bromo ketone. Below is the bromination of acetone.

The acid catalyzed bromination of a ketone. This occurs through the formation of an enol, which then behaves as a nucleophile and attacks the bromine. The reaction only occurs once, and leads to the formation of the monobromo ketone.
You should notice that the acid is regenerated in the last step of the reaction and is a catalyst. This means you can use a sub-stoichiometric quantity of a protic acid. The reaction is similar to the bromination of an alkene (see HERE) except the double bond of an enol is more reactive than that of an alkene. The lone pair of the oxygen atom is delocalized into the double bond making it more electron rich and hence more nucleophilic.
If only a single equivalent of bromine is added to the reaction it will give the monobromo ketone. If an excess of bromine is added, the reaction will eventually add a second bromine but it is a much slower reaction. The electron withdrawing bromine atom reduces the basicity of acetone slowing the first proton transfer. The presence of the bromine also slows the second proton transfer (and would encourage it to go on the other carbon atom of acetone). The bromine slows down the deprotonation as it destabilizes the positively charged transition state (electron withdrawing groups disfavor the build up of positive charge).

The positive charge of the oxonium species, the activated carbonyl, is spread over the substrate during the deprotonation. Unfortunately, the bromine is electron withdrawing and disfavors having a partial positive charge near it. This step slows down and it is easy to stop bromination after a single addition.
The second question you should be asking, is why can you perform the bromination in acetic acid? Why isn’t the carbonyl-containing acid also brominated in the α position? Acids and esters do not undergo tautomerization as readily as aldehydes and ketones. In acetone, about 1.7 x 10-7% of the enol is present. In ethyl acetate, this has fallen to < 1 x 10-15%. It effectively does not exist. The carbonyl group of an ester (or acid) is less polarized than the ketone as the second oxygen can delocalize its lone pair and feed additional electrons into the carbonyl group. It can also be argued that the ester carbonyl group is more stable than its ketone counterpart. Again, this is a result of the delocalization of electrons (think of the stability of alkenes increasing with substitution).
It is possible to synthesize α-bromo acids but this is achieved through an acyl bromide or chloride intermediate. Converting the acid to acyl halide activates the α-position. Now the carbonyl group is more polarized and enolization is easy; the halide is electron withdrawing making the carbonyl carbon more partially positive and there is little delocalization of the halide electrons. The acid is first converted to an acyl halide (acyl bromide normally uses PBr3, Br2, while acyl chloride uses SOCl2). Enolization of the acyl halide permits bromination. Finally, addition of water or an alcohol gives either the acid or an ester. Substitutions of carboxylic acid derivatives were covered HERE.

Carboxylic acids barely undergo enolization so they are inert to the standard conditions, but if they are converted into the far more reactive acyl halides first, then the reaction readily proceeds. The acid (or an ester or amide) can easily be obtained by reacting the acyl halide with the appropriate nucleophile.
Acetone, the ketone in the previous examples, is a symmetric ketone, and reaction can occur on either side of the carbonyl group to give the same product. If the ketone is non-symmetric, the two alkyl substitutents are different, regioselectivity becomes an issue as there are two possible products. This is shown with the bromination of butanone below:
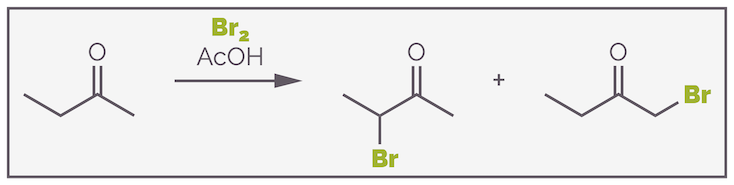
The reaction of a non-symmetric ketone can lead to the formation of two regioisomers.
Under acid-catalyzed conditions, the secondary bromide is formed (the first product above). The more substituted enol is more stable. This is the Zaitsev product and is formed for the same reason more substituted alkenes are more stable as discussed HERE. The more substituted enol is more stable due to hyperconjugation or σ-conjugation. When a full bonding orbital can overlap with the empty π* antibonding orbital of the C=C double bond, its electrons are effectively delocalized and this is more energetically favorable than localized electrons.
Alternatively, the transition state for the formation of the more substituted enol is lower in energy. As the cationic oxonium species is deprotonated there is a build up of positive charge across the molecule. Spreading this partial positive charge across the more substituted carbon leads to a more stable species, more substituted cations are more stable, and a lower energy transition state. This means formation of the more substituted enol is more favored as shown below:

Acid-catalyzed enol formation normally favors formation of the more substituted enol. The reaction is reversible and the more stable or substituted double bond is formed. Alternatively, the transition state leading to this regioisomer is lower in energy as the positive charge is spread over a more substituted system leading to increased stability.
The bromination will favor the secondary bromide.
Acid-catalysed bromination of a non-symmetrical ketone proceeds through the formation of the more stable enol. This corresponds to the Zaitsev product or more substituted alkene. This ultimately leads to the formation of the more substituted bromide.
Enolization is an equilibrium process that favors the formation of the most stable enol. As mentioned this is the more substituted enol. The less substituted enol is almost certainly formed and may even be formed faster as the proton is more accessible but this enol is less stable as there is less σ-delocalization. It readily reverts to the ketone. The more substituted enol is more stable. There will be a higher concentration of this enol and it will react with the bromine to give the product.
Keto-enol tautomerization allows aldehydes and ketones to react with other strong electrophiles. A method for the formation of diketones is the nitrosation of ketones, although I'll be honest and say this isn't the most common reaction (although I just read a thesis that made extensive use of it) but it makes good practice. An example is shown below:

The nitrosation of a ketone followed by hydrolysis to give a 1,2-diketone.
The first stage of this reaction is the formation of a highly electrophilic nitrosonium ion. Those of you that have encountered electrophilic aromatic substitution (HERE) will already know how this occurs, for the rest of you, it is a series of proton transfers that creates a good leaving group in the form of a hydronium ion or water. This is shown below:

The formation of the highly electrophilic nitrosonium ion.
The anionic nitrite anion is protonated by acid to form nitrous acid (a proton transfer). A second protonation occurs to create the hydronium ion, a good leaving group and a common motif in many reaction mechanisms. The leaving group is eliminated to give the highly electrophillic nitrosonium species.
Students often ask why is the oxygen of the OH group of nitrous acid is protonated, and neither the other oxygen or the nitrogen atom? There are two answers. The first is that all the atoms are protonated at some point; protonations are invariably fast, but reversible reactions. Organic chemists just draw the productive steps, chosing to ignore the rest. Secondly, you could argue that this oxygen is more basic than the rest; it is sp3 hybridized while the second oxygen has smaller sp2 hybridized orbitals for the lone pairs of electrons. The nitrogen is also sp2 hybridized, and is attached to electronegative oxygen atoms. But I prefer the first argument.
Under the acidic conditions required to synthesize the electrophile, a nucleophilic enol is formed, and these two react. The enol attacks the nitrosonium species at the nitrogen so that the electrons flow towards the electronegative oxygen atom. This gives the protonated nitroso-ketone. Deprotonation gives the initial product.

Formation of a nitroso-ketone by the nucleophilic addition of an enol to a nitrosonium ion.
Before hydrolysis of the nitroso group occurs it must undergo tautomerization to give an oxime. This shows that tautomerization is not specific to the carbonyl group and can occur with other functional groups, it simply describes the movement of a hydrogen within one molecule (and invariably occurs with the shift of a multiple bond). In this case, the nitroso compound is unstable and the tautomer, the oxime is stable. The stability is aided by internal hydrogen bonding.

Tautomerization of the nitroso compound gives an oxime that is hydrolysed under acidic conditions.
Hydrolysis of the oxime is the reverse of the condensation reactions covered in a summary on carbonyl chemistry HERE. The first step is protonation of the oxime to give a highly electrophilic cationic species. Water acts as a nucleophile and attacks to give an hemiaminal-like species as the tetrahedral intermediate. In this example, I've drawn an internal proton transfer as I'm lazy. I don’t like this version of the mechanism, and should have drawn the two-step process involving an external base and a proton source (but the drawings take the longest time and this summary is about the enol and this is just one long aside so I decided to be lazy). The protonation of the basic nitrogen atom leads to an ammonium cation that is a good leaving group. This facilitates the collapse of the tetrahedral intermediate to give an oxonium species. A final proton transfer gives the product. If this doesn't make sense read THIS.
Enolate formation
Enols are normally considered mild nucleophiles meaning they are limited to reactions with strong electrophiles. With the exception of the aldol reaction and possibly conjugate additions, both of which will get their own summaries at some point, I don't want to cover anymore reactions of enols as carbon-based nucleophiles (I will need to cover O-silylation at some point). Instead, I want to look at making more powerful nucleophiles that will permit a wider range of reaction.
The clue to making a better nucleophile is hidden in plain-sight, and the mechanism of base-mediated enol formation. The negatively charged enolate, with its surfeit of non-bonding electrons, is a good nucleophile.

The formation of the anionic enolate. This species is a good nucleophile.
Enolate formation requires the removal of the α-proton to leave an anion. This is possible as the α-proton is relatively acidic. The ease of deprotonation or the removal of a proton is related to the stability of the resulting anion or conjugate base. I have covered how you can predict this stability based on structure of a molecule in a previous summary HERE. There are three factors that you need to consider for enolate formation. The first is delocalization. An anion is more stable if it is delocalized rather than centred on a single atom. In the case of an enolate you can draw two resonance structures, one with the negative charge on the carbon and another with it on the oxygen atom. Remember, that resonance shows extremes and that the electrons are spread over the three atoms. Neither representation is correct (or incorrect), they are both required to show the real structure.
The second factor involves the electronegative oxygen atom. A charged conjugate base is more stable or favorable if the charge is on an electronegative atom. In this case, one of the resonance structures places the anion on oxygen. This resonance structure will have a bigger contribution to the real structure (the resonance hybrid) and leads to greater stability. It is possible to form this resonance structure directly by deprotoning the enol rather than the α-proton of the ketone.
Finally, the inductive effective helps stabilize the α-anion. The partial positive charge on the carbon of carbonyl draw electrons towards it spreading the charge.

It is possible to remove the α-proton next to a carbonyl group as the resulting anion, or conjugate base, is relatively stable. The anion is delocalized over three atoms. It mostly resides on an electronegative oxygen atom and the inductive effect makes the carbanion resonance structure more stable than might otherwise be the case.
These three factors work in harmony to lower the acidity of the α-proton and make it easy to remove. This can be seen if you compare the pKa of other C–H bonds as shown below:

The ease of deprotonation is influenced by a number of factors including delocalization and electronegativity. The α-proton is more acidic than in an alkane due to delocalization and the possibility that the resulting anion can reside on an electronegative atom. The more electronegative atoms or the more delocalization, the more stable the anion (conjugate base) and the easier the deprotonation is.
Deprotonation of a simple alkane is hard. The resulting anion is not stabilized and, as a result, is highly basic or unstable. The pKa of ethane reflects this, at around 50, it indicates that the hydrogen is non-acidic. Acetone on the other hand, with a pKa of 19, is readily deprotonated. The resulting anion is stabilized as described above. If you increase both the amount of delocalization and add a second electronegative heteroatom then the anion can be made very stable. The pKa of acetylacetone is 9, the proton is more acidic, or more readily removed than that of water.
General Reaction
Enolates are normally carbon nucleophiles, reacting to give a new carbonyl-containing compound as shown in the reaction below:
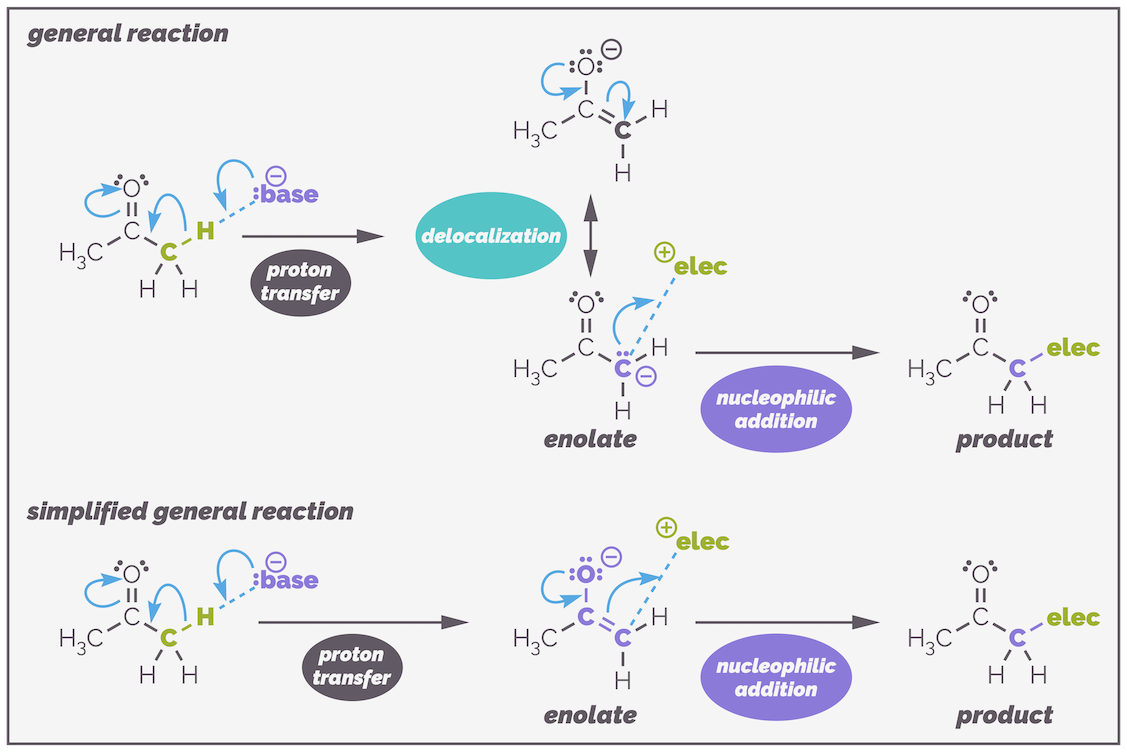
The general reaction of enolates. Deprotonation gives the enolate anion. This is resonance stabilized. The resonance structure with the anion on the oxygen makes a bigger contribution to the structure of an enolate but the enolate normally reacts through the carbon anion. The top version of the reaction shows the two structures an uses the lone pair of electrons on the carbon to react with electrophiles. Most chemists are too lazy to draw the two resonance structures every time and will use the lower simplification. The enolate is drawn as the more stable resonance structure and then the electrons flow through the enolate so that reaction occurs at carbon. Occasionally, you will see the reaction drawn showing only the carbon anion and sometimes you will see the wonderful atom specific curly arrow being used to show the α-carbon reacts.
The two resonance structures show that enolates are electron rich on both the α-carbon and the oxygen atom. This is indicated by the negative charge being shown on either atom. Both atoms can be the nucleophile but the majority of the reactions occur on carbon. Why? There are a number of ways you can rationalize this selectivity. Firstly (and a little simplistically), the negative charge is most stable on the electronegative oxygen atom. This means the oxygen is less reactive and reaction occurs through the less stable, more reactive carbon. A second argument is that the carbonyl bond is stronger than the C=C bond of the enol and this favors formation of the product that retains the C=O bond.
A more in-depth answer than most undergraduates require involves looking at the valence bond (frontier) orbitals. Reactions occur through the interaction of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The nucleophile, as the electron donor, will use the electrons of the HOMO. The drawing below shows the HOMO and LUMO of the allyl anion and an enolate. The former is drawn as it is simpler (so is a good starting point).

The frontier orbitals for the allyl anion and an enolate. The allyl anion only contains carbon and is symmetrical, the two ends of the system are the same. The enolate contains an electronegative oxygen atom and this distorts the orbitals. The electrons are pulled towards the oxygen atom meaning there is a large orbital on the π bonding orbital.
The allyl anion is symmetrical and the orbitals are easily described with the “electron in a box” representation. The C=C π bonding orbital has no node, with the electrons spread across the three atoms. The non-bonding orbital has a single node with the electron density concentrated on the terminal carbon atoms. This is the HOMO orbital and is the reactive nucleophilic component of the system. This means the allyl anion reacts at either terminal carbon but not the central carbon. Substituting one oxygen atom for a carbon causes a number of changes. Oxygen is more electronegative and stabilizes the negative charge. This means all the filled orbital will be lower in energy than the allyl anion and I should have drawn them lower to represent this! It also means the orbitals are distorted. As the oxygen has a lower energy atomic orbital (being more electronegative) it contributes more to the lower energy π orbital. The orbital is biggest on the oxygen. In the non-bonding orbital the situation is reversed. Now the biggest coefficient is on the carbon. This means the enolate is like the allyl anion in the sense that electrons are spread over the three atoms and that the greatest concentration is on the terminal atoms, the oxygen and the α-carbon. The difference is that the HOMO, the reactive nucleophilic orbital has a greater electron density on the α carbon and so reaction takes place here (it is not quite this simple, some reactions still take place on oxygen as it has a greater density overall, you then need to start considering hard and soft acids and bases but that is topic for another day).
Enolates tend to react at the carbon atom (and for this summary will only react at the carbon atom), so why do I draw them with the negative charge on the oxygen and then use two curly arrows to attack an electrophile? Why not just draw the carbon-centered anion and use a single curly arrow? Well, you will see that representation in some books and it is great. It is just using the most useful resonance structure. Personally, I prefer to draw the most favored (contributing the most) resonance structure. I should then draw the two curly arrows that connect this to the second resonance structure and then use that second resonance structure to attack the electrophile. But that is just too much like hard work, so the simplified version I will use as the oxygen-centered anion reforming the carbonyl group and causing the double bond to attack the electrophile through the α-carbon. Clear?
The simplest reaction of an enolate is protonation. This reaction returns the carbonyl group. I have already drawn it when discussing base-catalysed enol formation but here it is again so that you can practice these reactions:

Enolates are nucleophiles so will readily react with sources of relatively acidic protons.
In the first step, deprotonation of the α-carbon leads to formation of the enolate. This will react with a suitable electrophile, such as water, in a substitution reaction to give the carbonyl compound back.
Haloform Reaction
Enolates will also react with halogens such as bromine, but the reaction is very different to that of the enols shown earlier. Now a carboxylate anion will be formed along with a haloform (CHX3) in what is, unsurprisingly, called the haloform reaction. Neutralization of the carboxylate with acid leads to a carboxylic acid.

Base-mediated bromination of a ketone leads to the haloform (in this case, bromoform) reaction in which a C–C bond is cleaved.
The first part of the reaction proceeds as you might expect, the base causes deprotonation to give an enolate. This reacts with the bromine to form the α-bromoketone. The reaction is not catalytic in base with the base being consumed (converted to water). The bromide ion is not a good base so cannot deprotonate water.

The first stage of the bromoform reaction is exactly as expected, the base deprotonates the ketone to give an enolate that then participates in nucleophilic substitution.
The reaction does not stop at this point. By adding a bromine atom, you have added an electron withdrawing group. The two remaining protons on the methylene group are more acidic than the original methyl protons. They are more acidic as the resulting conjugate base is not only stabilized by delocalization as an enolate but there is also a strong inductive effect from the bromine atom. The α-bromoketone forms an enolate faster than the original ketone, and this reacts with more bromine to give a dibromoketone.

The α-bromoketone formed in the first stage of the reaction is more acidic than the starting material due to the electronegative bromine atom. It is more readily deprotonated to give a new enolate that can be brominated a second time.
The reaction doesn’t stop here either. The remaining proton is even more acidic than either the starting material or the α-bromoketone intermediate. The proton is next to both a ketone and two bromine atoms. It is deprotonated even quicker to form yet another enolate. Again, this reacts with the bromine to form a tribromoketone.

The dibromoketone is even more reactive than either the starting material or the intermediate. It has two electronegative bromine atoms that can enhance the acidicity of the proton on the α-carbon. As a result, the proton is easily removed to give yet another enolate that then reacts as a nucleophile to give the tribromide.
The reaction still isn’t finished. There are no acidic protons left so the hydroxide base behaves as a nucleophile and attacks the carbonyl group instead. This is an example of nucleophilic addition to a ketone. The resulting tetrahedral intermediate is not stable. It collapses to reform the carbonyl group and kicks out a good leaving group. This is Br3C–. Normally, you wouldn’t think of a carbanion as being a good leaving group, carbanions are very basic, meaning they are unstable. Yet in this case it is a good leaving group as the negative charge is stabilized by three electronegative bromine atoms. This leads to the formation of a carboxylic acid and a carbanion. While this carbanion is a good leaving group, it is still basic and it will deprotonate the acid to give bromoform (Br3CH) and a carboxylate anion. The carboxylate anion is favored as the negative charge is delocalized and can reside on two electronegative oxygen atoms.

Once the α-protons have reacted, the hydroxide anion acts as a nucleophile and attacks the carbonyl group to give a tetrahedral intermediate. This collapses expelling the good leaving group the tribrmo-stabilized carbanion. This gives a carboxylic acid and a carbanion that react to form bromoform and a carboxylate anion. The reaction has finished. If you want to isolate the carboxylic acid you need to neutralize the reaction by adding an acid.
This the end of the reaction unless you want to add some acid to neutralize the carboxylate and isolate the carboxylic acid.
A common question at this point is why does the hydroxide base attack the carbonyl group in this reaction but none of the previous examples? Or, why does it form the enolate when we want it to but the tetrahedral intermediate the rest of the time? Well the answer is that it does attack the carbonyl group all the time. It’s just that this reaction is normally unproductive but reversible. Attack leads to a tetrahedral intermediate but there is no leaving group and so when it collapses back to reform the starting ketone it kicks out the hydroxide anion and you are back to where you started. Equilibrium and the reversibility of reactions is very important. You can read about HERE.

Hydroxide anion will and does attack a carbonyl group. The reaction is normally unproductive as there are no suitable leaving groups so it just reverses and returns the starting materials. In the haloform reaction there is a different leaving group and hence you observe a different reaction.
Alkylation (and enolate formation part 2)
An important reaction of enolates is alkylation and the formation of C–C bonds. The generalization of this reaction (below) makes this look like a simple extension of the chemistry described above, but it is not. The choice of base becomes very important as you shall see.

General reaction equation for the alkylation of an enolate with a haloalkane. The first part of the reaction is deprotonation of the α-proton to form the enolate. The enolate then attacks a haloalkane in an SN2 substitution reaction to give the desired product.
A dicarbonyl compound, such as the β-keto ester below, can be treated with an alkoxide base, EtO-, to form an enolate that will readily undergo alkylation with an alkyl halide by an SN2 mechanism.
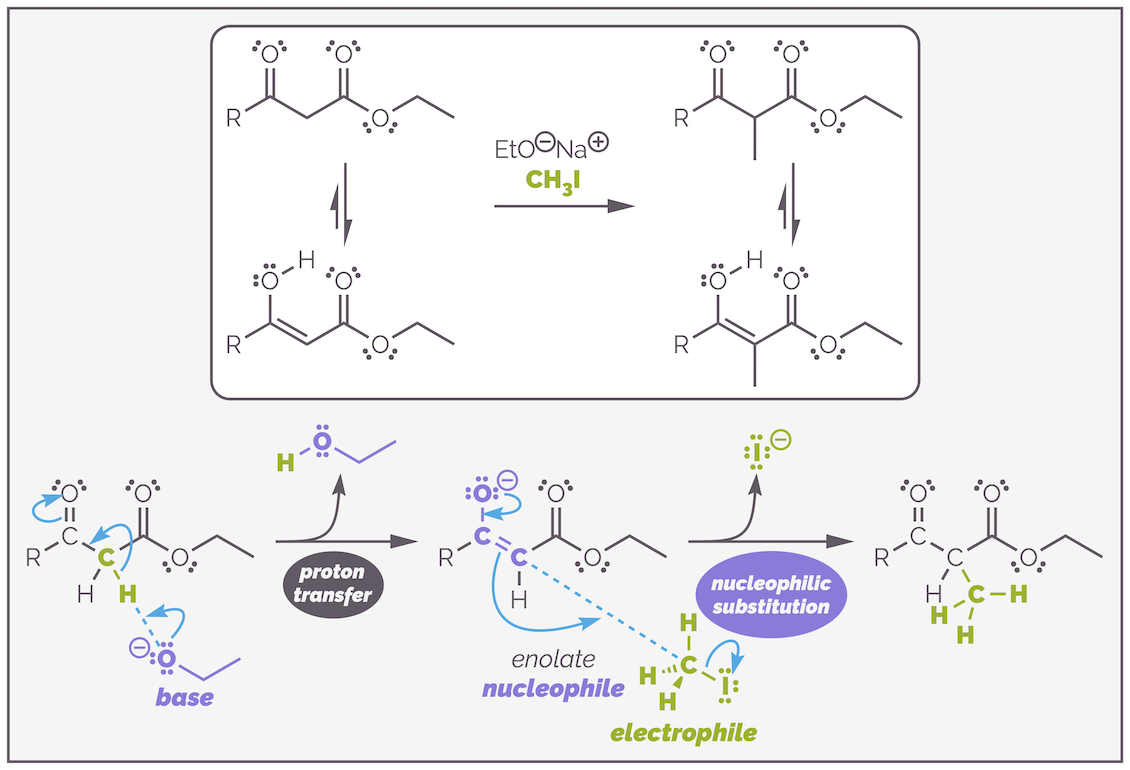
The alkylation of a β-keto ester. Enolate formation is relatively easy and can be achieved with sodium ethoxide. The choice of base is very important as you shall see. Otherwise the reaction follows the standard steps of deprotonation to give the nucleophilic enolate followed by nucleophilic substitution.
This reaction is only possible because of the two carbonyl groups. This lowers the pKa of the α-proton to approximately 11. This is lower than the pKa of the conjugate acid of the alkoxide (or pKaH), which is around 16 (the pKa of ethanol). This means the base is sufficiently strong to deprotonate the β-ketoester to create the nucleophile. The choice of alkoxide is important, but possibly not for the reason you are thinking. The alkoxide can also act as a nucleophile and attack the ester leading to transesterification. If you used a different alkoxide, such as CH3O-, you would get a mixture of methyl and ethyl esters. By using ethoxide as the base, you never notice that acyl substitution can also occur.
Choosing the wrong alkoxide base can lead to transesterification and formation of a mixture of esters (as well as alkylated compounds). In this example, the methoxide base was used instead of ethoxide and, as a result, a mixture of the desired ethyl ester and methyl ester are formed. The mechanism for the formation of the undesired methyl ester is shown.
Would this reaction work with a simple ketone such as acetone? No. The α-protons are not acidic enough. The pKa of acetone is 19 making its conjugate base a stronger base than an alkoxide. The enolate is barely formed as it has a greater hold on the proton. Mix the ketone, base and an alkyl halide together and the alkoxide will react with alkyl halide causing either substitution (ether formation) or elimination (alkene formation) depending on the structure of the alkyl halide.
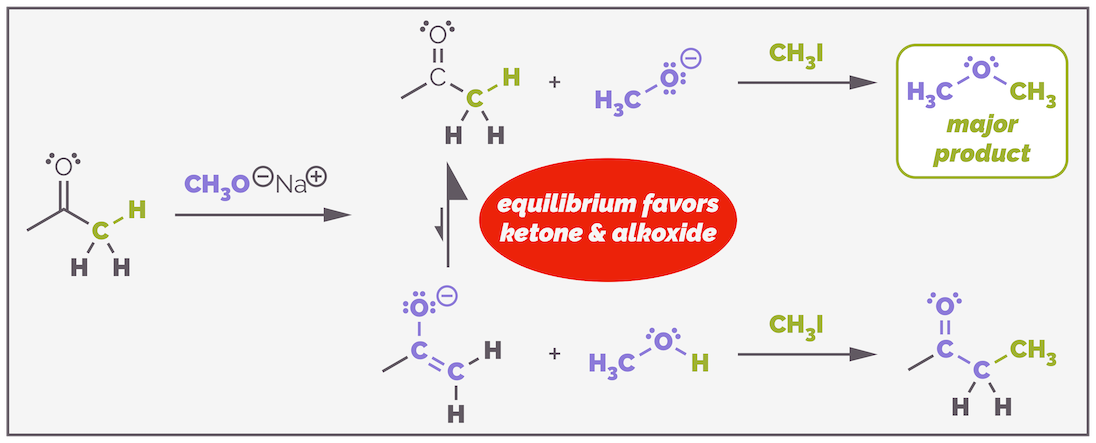
Alkoxides are not sufficiently strong bases to effectively deprotonate ketones and form a high concentration of enolate. The excess base left in the equilibrium mixture will react as either a base or a nucleophile with the alkyl halides leading to ethers and/or alkenes (depending on the structure of the alkyl halide).
There are a number of solutions to this issue. The first is to use the β-ketoester above. The increased acidity of the central α-protons leads to selective deprotonation. The enolate reacts efficiently in the alkylation reaction and then it is possible to remove the unwanted ester group. This can be achieved in a two step process involving hydrolysis to give the carboxylic acid followed by heating to bring about decarboxylation. The latter step requires the breaking of a C-C bond. This is normally a hard process but the presence of the ketone allows the reaction to proceed through a six-membered transition state (the more organic chemistry you do the more common this motif becomes. Nature loves a six-membered transition state) leading to carbon dioxide and an enol. Tautomerization gives you your ketone. Without the second carbonyl group, decarboxylation requires harsh conditions or special reactions.

One method of controlling regioselectivity and being able to utilize a weak base to form an enolate is to use a β-keto ester. These compounds are readily deprotonated and reliably undergo alkylation. The ester group can be removed through a process of hydrolysis followed by decarboxylation. Normally, the latter step is challenging and requires harsh conditions but the presence of a β-keto group allows the reaction to occur under milder conditions. A six-membered transition state involving the movement of six electrons allows the formation of carbon dioxide and an enol.
The alternative solution is to use a stronger base that can directly deprotonate the ketone and give you the desired enolate. The common choice for this task is lithium diisopropylamide or LDA. This is a strong base with a pKa around 35. This is so high as the negative charge is on a nitrogen atom, which is less electronegative than all an oxygen base, and is less stabilized. The two isopropyl groups further destablize the negative charge as they are electron donating but, more importantly, these groups are very bulky and they prevent LDA from behaving as a nucleophile. It only acts as a base. The resulting lithium enolate is still very reactive and can be used in alkylation reactions.

Lithium diisopropylamide is a strong, non-nucleophilic base that is often used to deprotonate ketones, esters and amides to form enolates. The mechanism is shown at the bottom of the diagram. I've drawn the deprotonation as a six-membered transition state with the movement of six electrons (three curly arrows) just to show how common this motif is (there is a version of the nucleophilic substitution that also involves a six-membered transition state to give the product and lithium iodide). Lithium amides are very reactive and normally are kept at -78 °C until the electrophile is added.
This chemistry works for a range of carbonyl containing compounds, including esters and amides, both functional groups that are harder to enolize than ketones. It should be noted that LDA doesn’t give good results with aldehydes, frequently leading to self condensation through the aldol reaction. I’ll cover the aldol reaction another time as it is very useful reaction (unless you want to alkylate an aldehyde). Below is a series of more successful examples:
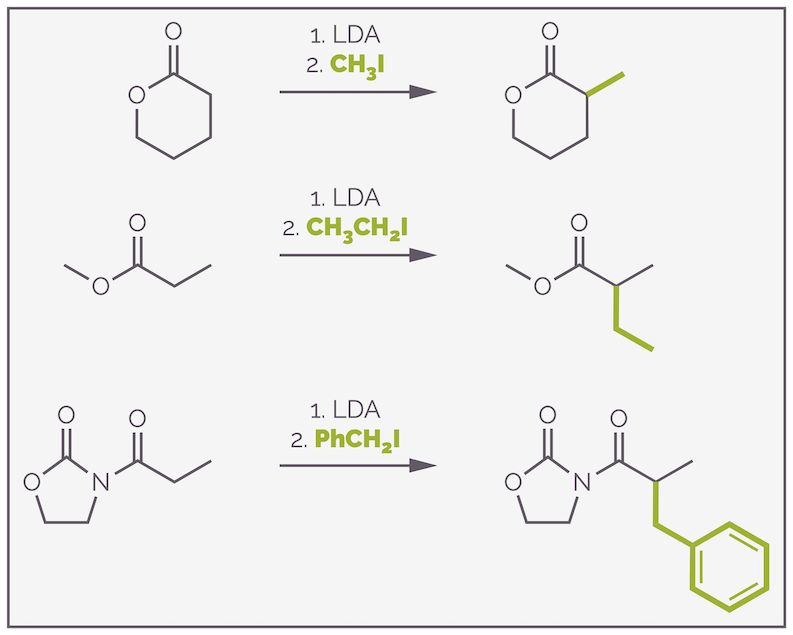
Examples of alkylation that can be achieved by the formation of a lithium enolate using LDA.
There is one last issue that needs to be addressed, what happens if the ketone is not symmetrical? Now there are two possible enolates so what is the regioselectivity or where does the alkylation take place? Enolates are effectively substituted alkenes, and, like alkenes, the more substituents the more stable the enolate. This means the top enolate (in the diagram below) is the more stable enolate. It is known as the thermodynamic enolate (it is effectively the Zaitsev enolate). The bottom enolate is formed faster as there are more protons (it is more statistically likely to form), the protons are more acidic (there are fewer electron donating groups destabilizing the carbanion), and the protons are more accessible (so react faster). This means the bottom enolate is the kinetic enolate.

A non-symmetrical ketone can lead to two different regioisomers of enolate. The more stable, more substituted enolate is known as the thermodynamic enolate while the enolate that is formed faster is called the kinetic enolate.
Formation of the kinetic enolate is more common and more easily achieved (formation of the thermodynamic enolate requires the establishment of an equilibrium and this frequently leads to a mixture of compounds). LDA is a bulky base, and it favors attack of the least hindered protons. This leads to the kinetic enolate. As long as you prevent equilibrium by maintaining a low temperature, normally –78 °C, using excess base to ensure full deprotonation, and not leaving the reaction for a long time then it is possible to get good yields of the desired compound.

The kinetic enolate can be formed by using a strong, bulky base in excess. The reactions are conducted at low temperature to avoid equilibrium and to favor removal of the more accessible protons. The reactions are performed quickly.
The thermodynamic enolate can be formed by doing the opposite! Use less than one equivalent of a small base at higher temperatures allows an equilibrium to be established, and this will often favor the more substituted enolate. There are a number of other methods used to prepare the thermodynamic enolate and I’ll discuss these in other summaries.

The thermodynamic enolate can be formed by performing the reaction under conditions that are likely to allow an equilibrium to be established. This includes using sub-stoichiometric quantities of small (often weak) bases, at higher temperatures and leave the enolate for long lengths of time.
Conclusion
Aldehydes and ketones are often in equilibrium with their enol form. The carbonyl compound undergoes tautomerization or enolization, where a proton is transferred from the α-carbon to the oxygen atom and the C=O π bond shifts to form a C=C double bond. Normally, this equilibrium favors the aldehyde or ketone to such an extend that the enol is not observable but this is not always the case. Compounds with a β-carbonyl group often favor the enol form. Formation of the enol can be either base or acid-catalyzed. Invariably, the thermodynamically more stable, more substituted or Zaitsev enol is formed. Enols are nucleophiles and can react at both the oxygen or carbon atom. The latter is more common (and I haven’t discussed the former in this summary). Enols react with strong electrophiles such as halogens or the nitrosonium ion to give α-haloketones and aldehydes or dicarbonyl compounds.
Enolates are the deprotonated version of an enol. They are formed by treatment of carbonyl-containing compounds with stronger bases. The negative charge makes them more nucleophilic than enols and they undergo a wider range of reactions. They react with halogens in the haloform reaction which leads to the breaking of a C–C bond and formation of a carboxylate anion. They also undergo alkylation. The regioselectivity of enolate formation can be controlled. The less substituted enolate, the kinetic enolate, can be formed by reaction with an excess of strong, bulky base at low temperatures. The more stable, more substituted enolate can be formed by establishing an equilibrium by treating the ketone with a small or weak base at room temperature or higher. Equilibrium requires a proton source so either use a weak base or less than one equivalent of base so that unreacted ketone can act as a proton source.
The chemistry discussed in this summary is only the start of a huge and vital area of synthetic chemistry (this summary is already far too long). Enols, enolates and their equivalents (compounds that behave as if they were either an enol or enolate) are among the most important functional groups for the creation of new molecules (even Nature uses enols and enolates to make molecules). There are more reactions of enols and enolates that you will need to cover, in particular the aldol reaction (and all its variants). Then there is the discussion concerning regio- and stereo-selectivity, which could take several summaries on their own. Finally, I haven’t mentioned all the enol/enolate equivalents such as silyl enol ethers and enamines. But hopefully, this summary has proven a useful introduction to this fascinating subject.