Eliminations
Introduction
The previous summary discussed substitution reactions, where a leaving group is exchanged for a nucleophile through one of two, often competing, mechanisms. In these reactions, the reagent acts as a nucleophile and attacks a carbon atom of the substrate to replace the leaving group. Many nucleophiles can also act as bases, and attack a proton or C–H bond. When this proton is on the carbon next to the leaving group (α to the leaving group) an elimination reaction occurs and an alkene is formed.

A secondary bromoalkane could react with hydroxide to give either an alcohol, by a substitution reaction, or an alkene by an elimination reaction.
Just like substitution reactions, eliminations can occur by different mechanisms depending on the substrate and the conditions. This summary discusses the two most common, E1 and E2 eliminations. At the end of the summary, I will give some hints on how determine whether a reaction is likely to be substitution or elimination and which of the four possible mechanisms is probably operating.
Elimination Reactions
Alkenes can be formed from substrates that have a leaving group and an α-proton, this latter term means a C–H bond on the carbon adjacent to the carbon with the leaving group. This is shown in the diagram below (but ignore the conformation, this diagram just shows the process, the details are in the summary):

The elimination of a leaving group (LG) and an adjacent proton (a proton in the α-position) leads to the formation of an alkene. This diagram is bad as it suggests the two groups being eliminated are on the same side of the molecule. As you will see in the coming sections this is not true (well there are examples of syn eliminations but these are rare).
Elimination commonly occurs by one of two mechanisms, E1 or E2 elimination (there are other elimination reactions but I’m discussing those that generally look like the transformation above, and appear at the start of undergraduate courses). Like the analogous substitution reactions, SN1 and SN2, these mechanisms differ by the number of reactants in the rate determining step (RDS). E1 reactions are first order or unimolecular, and only the substrate is found in the rate equation. E2 eliminations are second order or bimolecular and the rate is controlled by the interaction of both the substrate and the base. Ultimately, these differences relate to the timing of the lose of the leaving group and I'll discuss each in turn.
E2 Eliminations
The mechanism of an E2 elimination involves the base attacking a hydrogen atom. Donation of two electrons to form a bond removes the proton from the substrate and the electrons of the C–H bond create a π bond with simultaneous lose of the leaving group. All the bonds are made and broken at the same time in a concerted reaction. The reaction is second order or bimoleculecular with the rate equation dependent on the concentration of both the substrate and the base. There is one very important transition state in which all the reactants are aligned with the correct conformation. This is all summarized in the diagram below:
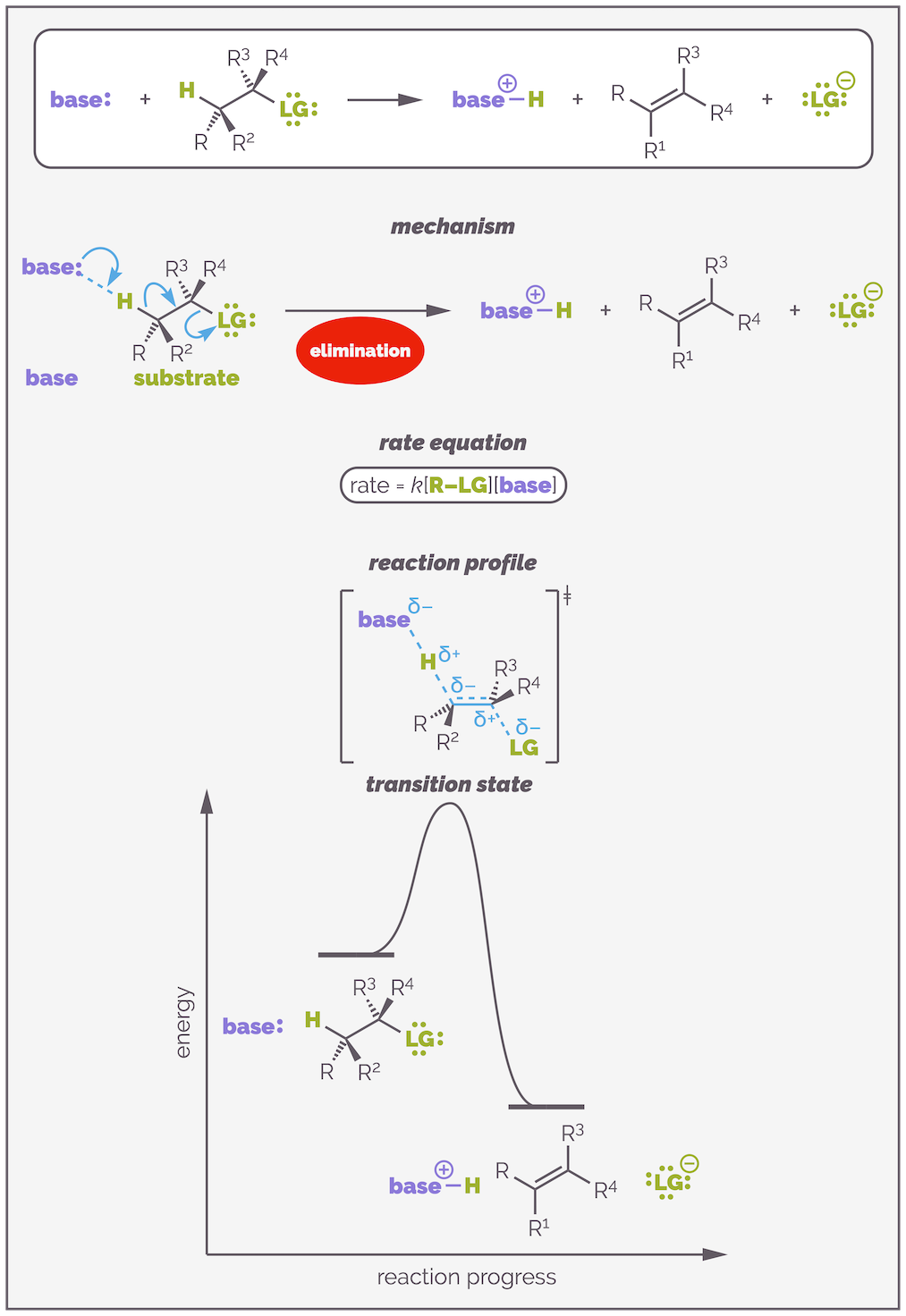
E2 elimination involves the reaction of a base and a substrate to create an alkene. It is a concerted process with all the bonds being made and broken at the same time. This means the rate equation involves both substrate and base. The reaction occurs in a single step and there is a single transition state that has the base, proton and substrate aligned.
E2 elimination only occurs when the hydrogen being removed and the leaving group are antiperiplanar to each other (there are examples of synperiplanar eliminations but these are rarer and normally by an Ei mechanism). This is shown in both the skeletal representation and Newman projection below. The easiest way to draw this requirement in the normal skeletal or line diagram representation is to rotate the carbon atoms so that both the C–H bond and the C–LG bond are in the plane of the paper or screen, and then make sure they are on opposite sides of the connecting C–C bond. The Newman projection is really helpful (for once) as it allows the planar relationship between the key bonds to be readily visualized. It also makes the relationship between the remaining substituents very clear, which will be important in the next paragraph.

There is a conformational requirement for E2 elimination. The hydrogen being removed and the leaving group must be antiperiplanar to each other. This is shown in the skeletal form and the Newman projection for the same molecule.
When there is a choice of protons to be eliminated, E2 eliminations are stereoselective favoring formation of the more stable, less hindered, E or trans alkene over the less stable Z or cis alkene. In the example below, three alkenes can be formed. The two internal alkenes account for the majority of the product and I'll ignore the terminal alkene (in brackets) for the time being (that’s a discussion on regioselectivity and comes later).

E2 eliminations can be stereoselective if there is a choice of protons that can be eliminated. Invariably, the more stable geometry of alkene will be formed (by more stable, I mean the one that places the largest substituents on opposite sides of the alkene).
The reaction is referred to as stereoselective as the reaction mechanism allows two different stereoisomers to be formed and yet one is preferred. You can understand the selectivity for the trans alkene by inspecting the Newman projection of the two potential transition states (shown on the reaction profile below). Both transition states have a different hydrogen atom antiperiplanar to the bromine atom. When the purple proton is eliminated, the transition state will be higher in energy as there will be steric interactions between the two methyl groups. This transition state is disfavored. Elimination of the green proton occurs through a transition state that places the methyl groups as far a part as possible. It is favored and the reaction predominantly forms the E or trans isomer.

When there is a choice of protons that can be eliminated, E2 reactions progress through the transition state that has less steric interactions between substituents.
If there is only a single proton that can be removed, E2 eliminations are stereospecific. The mechanism of a stereospecific reaction allow the formation of a single stereoisomer. SN2 reactions are stereospecific as they must occur with inversion of stereochemistry, SN1 reactions are not as they can occur with inversion or retention (they can be 100% stereoselective but they are not stereospecific ... terminology can be confusing sometimes). This is demonstrated if you look at the E2 elimination of the two diastereomeric bromides below:

E2 eliminations are stereospecific if there is a single proton that can be removed. This means that two diastereomers will give to different geometric alkenes under the same reaction conditions. This is due to the requirement for the proton and the leaving group to be antiperiplanar to each other.
In each case, there is a single proton α to the leaving group. This means that for each diastereomer there is a single conformation that places the proton antiperiplanar to the leaving group. Only when the substrates adopt these conformations will they undergo E2 elimination. As a result, each diastereomer can only deliver a single alkene. The top reaction will be faster than the bottom one. In the top reaction, the two bulky phenyl groups are opposite each other. It is easy for the substrate to possess this conformation. The reactive conformation for diastereomer 2 has the two phenyl groups next to each other. This is unfavorable and most of the population of diastereomer 2 is in a different conformation. It is slow to react.
The necessity for an antiperiplanar arrangement of proton and leaving group has an interesting effect on the chemistry of cyclohexane derivatives. A substituent on a cyclohexane ring can can either be equatorial or axial. The two conformations are in equilibrium with each through a process called ring-flipping, which is simply the rotation of C–C bonds. If you have no idea what I'm talking about, read this summary HERE.

The chair conformation of a cyclohexane ring places a substituent in one of two positions; it is either equatorial or axial. A change in conformation, known as ring flipping, allows the substituent to swap back and forth between these two positions. When a substituent is equatorial, the only bonds antiperiplanar to it are C–C bonds. These cannot participate in E2 elimination. When the substituent is axial, there are potentially two antiperiplanar C–H bonds. This means only an axial leaving group can participate in E2 elimination.
If the substituent (our leaving group) is in the equatorial position then there are only C–C bonds antiperiplanar to it. There are no protons antiperiplanar and E2 elimination is not possible. If the cyclohexane ring-flips, placing the leaving group in the axial position then two protons are now antiperiplanar protons. These can be removed in an E2 elimination to form an alkene. Simplistically, cyclohexanes can only undergo E2 elimination if both the leaving group and the proton at in axial positions.

Cyclohexane derivatives only undergo E2 elimination when both the leaving group and the proton to be removed are parallel to each other in an antiperiplanar conformation. This can only occur if they are both axial.
As with acyclic examples, this conformational requirement, the need for proton and leaving group to be axial, means diastereoisomers react at different rates and can give different products. This is demonstrated by the reaction of the menthol derivatives below. The top diastereoisomer gives two alkenes. In the most stable conformation of the substrate the large isopropyl group is equatorial, minimizing disfavorable 1,3-diaxial interactions. This forces the chlorine leaving group into an axial position. There is axial proton on each of the adjacent carbon atoms. E2 elimination could involve either and this leads to the two isomeric alkenes. I'll explain why one (in green or the left-hand side) is favored over the other later.

Diastereomeric cyclohexane derivatives can give different results in E2 eliminations. In the top example (diastereomer 1), the favored conformation has the isopropyl group in the equatorial position to minimize 1,3-diaxial interactions. This places the chlorine leaving group axial. There are two possible axial protons that can be removed during E2 elimination to give two different alkenes. In the bottom example (diastereomer 2), the favored conformation still has the isopropyl group equatorial but this places the chlorine leaving group in an equatorial position. Equatorial leaving groups cannot react by an E2 mechanism as there are no antiperiplanar protons that can be removed. Before this diastereomer reacts it has to ring flip to the less favored conformation. This takes energy and causes the activation barrier to be larger and slows the rate of reaction. When ring-flipping occurs and the chlorine is axial, there is only one antiperiplanar proton that can participate in the elimination processes and so only one alkene can form.
The second diastereomer (bottom of the diagram) only differs by the configuration of the chlorine leaving group. The most stable conformation places the large isopropyl group equatorial as before but this now forces the chlorine atom into an equatorial position (this isomer is more stable than the top isomer as all substituents are equatorial). There no protons antiperiplanar to the leaving group, only C–C bonds. In this conformation, diastereomer 2 cannot react by an E2 mechanism (I'm not saying it can't react, just that one pathway is closed off to it). Before it can participate in an E2 elimination it must ring flip to the disfavored conformation in which all the substituents are axial. Now there is a single axial antiperiplanar proton that can be removed. One alkene is possible. The reaction is much slower than the elimination of the first diastereomer as it takes energy to cause the ring flipping and this increases the activation barrier.
The first diastereomer above showed that there is another form of selectivity that you must consider and that is regioselectivity. This occurs when there is more than one proton that can be eliminated and resulting alkenes are in different positions or the products are isomers of each other. Regioselectivity describes where a reaction occurs (not what stereoisomer is formed).
A quick mention of how organic chemists describe alkenes. Alkenes can be classified by the number of substituents (non-hydrogen atoms) attached to the alkene. Below are the four different possibilities. Monosubstituted alkenes have a single substituent. Disubstituted alkenes have two and this continues up to four substituents.

Alkenes can be classified by the number of substituents (non-hydrogen atoms) attached to the double bond.
Normally, the more substituted alkene is more stable. The reason is beyond most introductory organic chemistry courses so skip to the next paragraph if you don't care for more detail. Of course, you wouldn't be reading this post if you weren't interested so here is slightly more explanation. In one word, hyperconjugation, in two words, σ-conjugation. If you overlap a full orbital with an empty orbital, you will create a new, more stable orbital. The overlap of two orbitals, as long as both aren’t full, leads to a lower energy combination that allows the electrons to be shared over a larger volume. This is the reason that tertiary carbocations are more stable than primary carbocations, there is electron delocalization into the empty 2p orbital. Alkenes are the same, if electrons can delocalize into the empty π* antibonding orbital, then a lower energy orbital is formed and the compound is more stable. The more substituents, the more C–H or C–C bonds can be parallel with the π* antibonding orbital and the more delocalization is observed.
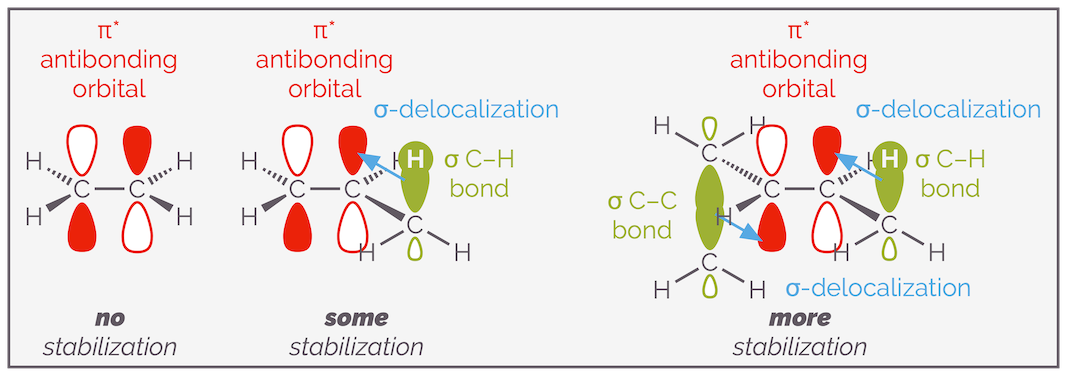
The more substituted a double bond the more stable it is (all other factors being equal). This is a result of the delocalization of the electrons of σ bonds into the π* antibonding orbital of the alkene.
The example below shows that two different alkenes can be formed (make sure you can see why it is only two and that there is neither another regioisomer nor a another stereoisomer that can be formed). The major product is the more stable internal alkene as it has more substituents. This product is sometimes called the Zaitsev product. The least substituted alkene is sometimes called the Hofmann product.

E2 elimination with a small (sterically non-demanding) base favors formation of the more stable, more substituted alkene. This is sometimes known as the Zaitsev product.
Changing the base can change the regioselectivity of E2 eliminations. Bulky, more sterically demanding bases, can favor the formation of the less substituted, Hofmann product. The size of base can hinder approach to the internal hydrogen slow due to interactions with the rest of the substrate. Removing the more acidic and more accessible methyl protons is a faster reaction and more of this product is formed.

With E2 eliminations, changing the base can influence the regioselectivity. A large bulky base is slower to react with the internal protons compared to the more acidic, more accessible, methyl protons. This change in rate allows the reaction to favor the formation of the less stable, less substituted, Hofmann product.
The characteristics of the leaving group can also influence regioselectivity in E2 eliminations. Good leaving groups, those with a weak C–LG bond, favor the formation of the predicted, more substituted, Zaitsev alkene. The proton is removed at the same time as the leaving group departs and this leads to a transition state that resembles the alkene. The more stable the alkene (the more substituted) the lower in energy of the transition state and the faster it will form.

A good leaving group favors the formation of the more substituted, Zaitsev product as the reaction proceeds through a transition state that resembles the alkene. The more stable the alkene the lower the activation barrier and the faster the products is formed.
Poor leaving groups, those that have a strong C–LG bond, favor the least substituted alkene or the Hofmann alkene. Again, the selectivity can be understood by considering the transition state. When the substrate has a poor leaving group, the base starts to remove the proton before the leaving group departs (this starts to change the mechanism to E1cB but that is a story for a different summary). This leads to a build up of negative charge (a partial charge, δ–) on a carbon atom. Less substituted carbanions are more stable than more substituted as less electron density is being pushed onto the carbon by the inductive effect or hyperconjugation.

A poor leaving group favors the formation of the less substituted alkene or the Hofmann product. With a poor leaving group there is a build up of negative charge as the proton is attacked earlier. The transition state resembles as anion and these are more stable with fewer electron donating substituents.
An aside: Valence bond or frontier orbitals
The description of elimination above is usually good enough for introductory undergraduate courses but I always find it a little unsatisfactory when there is no explanation of why E2 elimination requires an antiperiplanar arrangement of proton and leaving group. One explanation involves a simplified orbital view of the reaction.
When the base attacks the proton there is an overlap of the HOMO of base (often a lone pair of electrons in a non-bonding orbital) with the σ* antibonding orbital of the C–H bond. This leads to the C–H bond breaking. The electrons of the C–H σ bond overlap with the empty σ* antibonding orbital C–LG to create the new π bond and break the C–LG σ bond. The two orbitals must be in the same plane to form the new π orbital. There are two conformations of the molecule that allow this overlap, the antiperiplanar and synperiplanar arrangements. The latter is disfavored because it is a high energy eclipsing conformation while the former is a staggered conformation. The antiperiplanar conformation is favored because it has the bonds and hence orbitals parallel, maximizing the overlap of the orbitals.

E2 elimination occurs through the antiperiplanar conformation to maximize the overlap of the C–H σ bond and the C–LG σ* antibonding orbital. These mix to create the new π bond. The synperiplanar conformation is disfavor as it is eclipsed (high energy) and the orbitals are not parallel leading to poor overlap.
E1 Eliminations
Where E2 eliminations are bimolecular with both the substrate and the base in the rate determining step, E1 eliminations are unimolecular or first order reactions. The rate of elimination is dependent on only the concentration of the substrate and not the base. As a result, E1 eliminations are stepwise reactions that proceed by ionization to give a carbocation intermediate followed by deprotonation to form the alkene. All this information is summarized in the diagram below:

E1 elimination is a unimolecular reaction that is first order with respect to the substrate. This means it is a stepwise reaction, with the first step of the mechanism being ionization to form a carbocation intermediate. The second step is proton transfer which leads to the formation of the alkene. The reaction profile for the two step process is given at the bottom of the diagram.
You should see a striking similarity to SN1 substitution. Both reactions have the same first step, the ionization of the substrate to give a carbocation intermediate. This makes sense as the rate of reaction is controlled by the substrate only. The rate equation is the same for both reactions, the difference is the fate of the carbocation. The stability of the carbocation is important leading to tertiary leaving groups reacting faster than secondary or primary. The second step is where there is a difference, and this fast step is where the other reactant is involved. The reactant can determine whether the product is that of substitution or elimination. As the first step is the same, the two reactions nearly always compete with each other and it is common to observe a mixture of products.

Both E1 and SN1 have the first same step (ionization if you ignore the activation of the leaving group in this example) and share a common intermediate, the carbocation. The same substrates favor the dissociation pathway (E1 or SN1 versus E2 & SN2). The difference is the fate of the carbocation and it depends on many factors but an important one is whether the other reactant is a good nucleophile or not.
The reaction above shows emphasizes that both substitution and elimination proceed through the same intermediate. When a tertiary alcohol is treated with a strong acid, such as HBr or H2SO4, it is protonated to create a good leaving group. The hydronium ion dissociates and a carbocation is formed. When HBr is used, there is a good nucleophile present, and SN1 is possible with the bromide anion acting as a nucleophile and forming the tertiary bromide. When sulfuric acid is used the conjugate base, HSO4–, is a poor nucleophile and now E1 elimination is the predominant pathway. In both cases it is highly likely that a mixture of products will be formed.
There are no conformational requirements for the first step of E1 elimination. The leaving group departs and a carbocation is formed. Each of the bonds to the carbocation is free to rotate (as long as they are not part of a ring). The second step, elimination of the proton and formation of the alkene occurs when the C–H bond is parallel with the empty 2p orbital of the cation. This maximizes orbital overlap and allows formation of the alkene π bond.

E1 elimination proceeds by lose of the leaving group to give a carbocation that can free rotate. Removal of the proton and formation of the alkene requires the C–H bond to be parallel with the empty 2p orbital of the cation.
Formation of the cation and free rotation of the C–C bond to align the C–H bond means E1 elimination is not stereospecific, the mechanism can allow the formation of either geometry of alkene. The reaction is stereoselective and the more stable trans or E-alkene is normally favored as this places the largest substituents at either end of the alkene on opposite sides.
The stereoselectivity arises as the transition state leading to the trans or E-alkene is lower in energy due to the bulky substituents being far apart (if the reaction is reversible, dehydration giving the alkene and hydration giving the alcohol, then the selectivity will also be influenced by the stability of the alkene. The trans-alkene is more stable as the substituents don't interact). This is shown below. The key is that the C–C bond of the carbocation must rotate to orientate the C–H bond parallel with the empty 2p orbital. As it could rotate either clockwise or anticlockwise, it will favor the conformation that places the substituents far apart.

In E1 elimination, the only requirement is that the C–H bond of the proton being removed is parallel with the empty 2p orbital. Free rotation of the C–C bond in the cationic intermediate allows the species to adopt a conformation that minimizes steric interaction. This normally results in the E or trans-alkene being favored.
Theoretically, this means two diastereomers of a substrate should give the same alkene (unlike E2 elimination where they give different stereoisomers). I was unable to find an example of this reaction in the literature, and the following diagram should be taken with a pinch of salt. Experience suggests both would give a mixture of stereoisomers with a combination of kinetic and thermodynamic factors influencing the outcome depending on the exact reaction conditions.
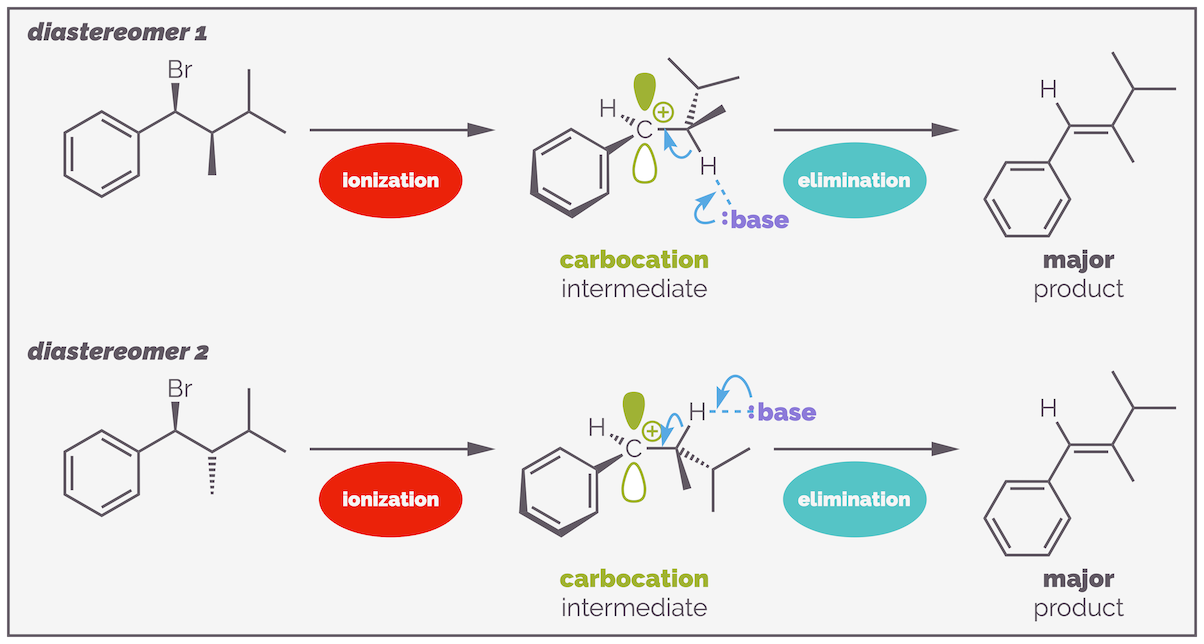
In theory, E1 eliminations should give the same stereoselectivity regardless of the initial diastereomer used. This probably doesn't happen in reality as there are multiple competing reaction pathways (as you will see) and, if I'm honest, I couldn't find an example that had experimental data provided. So take this as an illustrative example but not necessarily true.
E1 elimination normally gives the more stable, more substituted alkene, the so called Zaitsev product. The base has no effect on the regioselectivity of E1 elimination. Once the carbocation has been formed, virtually anything with a lone pair of electrons will cause deprotonation so alteration of the base is ineffectual. The more substituted alkene is formed fast as the transition state leading to the more stable alkene is lower in energy. The transition state has some double bond character so will have lower energy if it has more substituents for the same reason more substituted alkenes are more stable. I have also seen it argued that this transition state is more stable as the build up of positive charge is spread out of the more substituted carbon. These arguments more or less mean the same thing. Below is yet another reaction profile showing this.

E1 eliminations are frequently regioselective and will favor the formation of the more substituted alkene (the Hofmann product). The reason for this is that the transition state that leads to the more stable geometry of alkene normally has the lowest energy so the more substituted alkene is formed faster.
Selecting E1 versus E2 elimination
The two elimination mechanisms are in competition (because of course they are). This means they make popular questions in assessments. In reality, most reactions give mixtures and probably occur by a variety of mechanisms but it is possible to identify which reactions are likely to proceed by an E1 and which by an E2 mechanism. Like substitution reactions, the reaction pathway is influenced by a number of factors, with the main three being substrate structure, the base, and the solvent.
But, and here is the kicker, the discussion of E1 versus E2 is rather artificial, as they are also competing with the substitution reactions, and, as I will discuss later, the real competition is probably E1 versus SN1 and E2 versus SN2.
1. Substrate R–LG
The substrate is important when considering the mechanism as it appears in the rate equation for both pathways. As the rate always depends on the concentration of the substrate, a good leaving group, LG, will increase the rate of both E1 and E2 reactions.
All substrates (that can form an alkene by elimination) can undergo E2 elimination, making this reaction pathway more common than E1.

All substrates that can undergo elimination can undergo E2 elimination (I know this is a wild generalization that doesn't take into account mechanisms other than E1 & E2 but it is a reasonable starting point).
E1 elimination requires the formation of a relatively stable carbocation. This means only substrates that can stabilize the cation by either the inductive effect (tertiary leaving groups) or delocalization (α to an aromatic ring, double bond or heteroatom) can react through the E1 pathway. These substrates will often react by a mixture of E1 and E2. Any other substrate has to be E2.

An E1 pathway is only favored when a relatively stable carbocation can be formed.
A pictorial summary of which substrates can undergo E1 elimination is below:

All substrates with the appropriate arrangement of hydrogen and leaving group can undergo E2 elimination but only some of them can follow the E1 pathway. These have to be able to stabilize a carbocation.
2. The Base
The characteristics of the base, whether it is a strong or weak base, will only influence the rate of E2 elimination. It has no effect on E1 as the base does not appear in the rate equation for this mechanism.
A strong, non-nucleophilic base will increase the rate of E2 elimination and disfavor substitution or E1 elimination. Strong bases have weak conjugate acids with high values of pKaH. Anions are normally stronger bases than neutral compounds. I have a whole summary on comparing relative acidities and basicities HERE.

A rough scale of bases. Carbanions are very strong bases. Non-stabilized oxygen and nitrogen anions are strong bases. If they are stabilized by delocalization they are weak bases. Neutral species and the halides are very weak bases.
E1 elimination is a slow reaction. It requires the formation of a carbocation, it takes energy to break the C–LG bond and there is no compensation until a new bond is formed. No change in base will influence the rate of E1. If E2 elimination is also slow, then both mechanisms can compete. But if you use a strong base that accelerates E2 or you use a high concentration of base, which also accelerates E2 (base is in the rate equation) then E2 dominates.
3. The Solvent
The solvent can influence the mechanism by either stabilizing (or not) the carbocation intermediate, the leaving group, and solvating the base (decreasing the reactivity of the base). Polar protic solvents favor E1 elimination. Polar solvents can stabilize the cationic intermediate (or, more accurately, the transition state leading to the intermediate) and speed up ionization. Polar protic solvents disfavor E2 elimination as they can solvate the base and make it less reactive.

Solvent can influence the mechanism of the reaction by stabilizing the charged intermediates of E1 thus enhancing the rate of E1 elimination. Alternatively, it can solvate the base, making it less reactive and disfavoring E2 elimination.
Alternatively, polar aprotic solvents can slightly enhance E2 elimination. They are unable to solvate the base and, as it becomes more reactive, the rate of E2 can increase.
Solvent can play a major role in determine if a reaction is elimination or substitution, and then which of the four possible mechanisms as it greatly influences both leaving group ability (E1 & SN1) as well as basicity versus nucleophilicity. It can be complicated, and it is often better to understand the basics first before bringing this subject in!
Substitution versus Elimination
Most reactions can give a mixture of products. It is rare that a single reaction pathway operates in isolation and the four mechanisms I have covered, E1, E2, SN1 & SN2, compete with each other (along with a couple of mechanisms I haven't discussed). As both E1 & SN1 proceed through a carbocation, it is often stated that these two compete while E2 competes with SN2. While a useful generalization, it is not true. The reaction of tert-butyl bromide with either ethanol or ethoxide swaps between SN1 and E2 pathways:

The strength of the nucleophile or base can influence which mechanism is operating (and this can be influenced by the solvent).
The pathway of a reaction is influenced, as always, by the substrate, the reactant (strong/weak nucleophile/base), the solvent and the temperature (amongst other factors). These are not listed in any particular order and all factors influence the reaction pathway. Don’t get fixate on any one of the them (or the order below).
1. Substrate
A primary alkyl leaving group favors SN2 and possibly E2. Both SN1 and E1 are disfavored as the intermediate cation will not be stable (unless the leaving group is α to alkene, aryl group or heteroatom that can stabilize the cation).
Secondary substrates can react through any of the reaction pathways, and you are going to need to think about the reactant, the solvent or the temperature if you want to predict the mechanism. Yes, we all hate leaving groups at a secondary position.
Leaving groups at a tertiary position rarely participate in SN2 reactions (an exception can be found in the last summary HERE). But they can react through the other three mechanisms (SN1, E1 & E2). It is necessary to consider the other factors below to predict the predominant pathway.
The bigger and bulkier the substrate, the greater the possibility that it will undergo elimination (E1 or E2). The bigger the molecule that harder it is for a nucleophile to approach the carbon atom and it will always be slightly easier to pick a proton of the edge.

Generalization of effect of substrate structure on possible reaction pathway. These aren't rules but a good starting point.
- Primary alkyl leaving groups favor SN2 (and E2).
- Tertiary alkyl leaving groups can react by SN1, E1, and E2.
- Bigger and more sterically demanding substrates favor elimination.
2. Reactant - Strong or Weak Nucleophile or Base
In undergraduate courses, strong nucleophiles or bases are normally anions while weak reactants tend to be neutral. This is a generalization, and there are many exceptions, but it is a good starting point. Strong nucleophiles and bases encourage SN2 and E2.
Larger, more sterically demanding reactants are normally better bases than they are nucleophiles. It is easier for a bulky reactant to approach a small proton and behave as a base than it is for it to approach a carbon atom.

The size of the reagent can make a difference. Small reactants can easily approach the backside of the carbon and cause substitution. Small reactants are often good nucleophiles. As the reactant gets bigger, it is more sterically demanding and struggles to approach the carbon. It can still attack a small proton found on the outside of the molecule and larger reactants are often basic but not nucleophilic.
Annoyingly, many lecturers will, at one time or another, equate nucleophilicity and basicity, saying stronger bases are stronger nucleophiles and pKa can act as a good proxy to measure nucleophilicity. I'm guilty, having said this in lectures and on this website. It isn't true. pKa may offer a starting point but you must understand that nucleophilicity and basicity refer to two different properties (and not just because the reagents react with different atoms). Basicity measures a thermodynamic effect, it compares the stability of starting materials and products in an equilibrium process involving proton transfer. A strong base favors the lefthand side of the reaction, the protonated form, and will have a low high pKa. Nucleophilicity is a kinetic effect. It compares the rate of a reaction. A strong nucleophile reacts quickly, a poor nucleophile reacts slowly. This means there is no reason why basicity and nucleophilicity should match.
Solvents also have a huge effect the reactivity of nucleophiles and bases. Solvents are discussed in the next section.
Strong reactants, either strong nucleophiles or strong bases will favor SN2 and E2 pathways. The reactant is in the rate determining step (and the rate equation) and the stronger it is, the faster the reaction will be. Weak nucleophiles and bases do not promote SN2 or E2 reactions, and allow SN1 and E1 pathways to compete. No reactant actually enhances SN1 or E1, they only influence the rate of the bimolecular mechanisms.

The rough categorization of common reactants and their favored (but not only) reaction pathway.
Increasing the concentration of the reactant will favor SN2 and E2 reactions. The reactant appears in the rate equation for both reactions and increasing the concentration will increase the rate of both SN2 and E2. The reactant has no effect on the rate of SN1 and E1.
- Strong reactants favor SN2 or E2.
- Weak reactants allow SN1 and E1 to compete.
- Bulky reactants are often more basic than nucleophilic (encouraging E1 and E2).
- Increasing the concentration of the reaction favors SN2 and E2.
3. Solvent
The influence of solvent on the mechanistic pathway of a reaction cannot be over stated, it is very important, and yet it is the least discussed factor at undergraduate. This is understandable as its effects can be subtle and at the start of your chemistry career, there is a need to get to grips with the basics, such as substrate effects, before looking at the murky area of solvents.
I discussed solvents HERE when looking at substitution reactions but here is a very brief overview:
Polar protic solvents, such as water, alcohols and carboxylic acids, act as hydrogen bond donors to heteroatoms and anions, effectively forming a protective layer around the reactants and making them less reactive. This is known as solvation. Solvation is greater for smaller atoms as hydrogen bonding is stronger between atoms of similar (or at least closer) size.

Polar protic solvents favor E1 and SN1 pathways. They encourage ionization by solvating both the anion (leaving group) and carbocation. Hydrogen bonding allows them to solvate and reduce the activity of nucleophiles and bases, slowing both E2 and SN2. Polar protic solvents can enhance E2and SN2. They are poor at solvating the nucleophile or base and hence it is stronger or more reactive, which accelerates the two bimolecular mechanisms.
Polar aprotic solvents, like acetone, DMSO (dimethylsulfoxide), acetonitrile and DMF (dimethylformamide), are incapable of being hydrogen bond donors. They are poor at solvating anions and heteroatoms, and leave reactants ‘naked’ and more reactive.
Not all good nucleophiles are good bases and vice versa. As a rough guide, if you are comparing atoms in the same row of the periodic table nucleophilicity does follow basicity and as electronegativity increases so basicity drops (or pKaH decreases). The strength of a nucleophile does not necessarily follow the strength of a base down a group. Nucleophilicity down a group is strongly influenced by the solvent. In polar protic solvents, nucleophilicity does not follow basicity. The protic solvent can hydrogen bond to the anion, forming a protective layer around the anion making it less nucleophilic. Stronger hydrogen bonds are formed with smaller atoms. Those atoms at the top of a group have greaster solvation and are less reactive. Atoms at the bottom form weaker hydrogen bonds, they are less solvated and more nucleophilic. With polar protic solvents the strength of a nucleophile increases down the group while the basicity decreases. Polar aprotic solvents cannot donate a hydrogen bond to an anion so there is less interaction between the solvent and the anion. In this case, nucleophilicity normally mirrors basicity. Bottomline, solvent is important and can make a big difference.
Polar protic solvents favor SN1 and E1 reactions as the solvent can solvate both the anion and cation. This increases the rate of ionization and formation of the carbocation. Protic solvents disfavor SN2 and E2 as solvation of the nucleophile/base reduces the strength. Polar aprotic solvents are the opposite. These tend to favor SN2 and E2. They cannot stabilize the anion of the leaving group as well as protic solvents leading to less ionization and they cannot solvate the reactant as well, which encourages nucleophilic addition and/or deprotonation. My notes say that polar aprotic solvents favor SN2 over E2 but I can't formulate a good argument why!
- Polar protic solvents favor SN1 and E1.
- Polar aprotic solvents favour SN2 or E2.
4. Temperature
Increasing the temperature of a reaction favors elimination. Elimination requires more energy as more bonds are broken so raising the temperature allows E1 and E2 to compete with the substitution mechanisms.
A useful simplification is if you consider the change in Gibbs free energy of a reaction. This is discussed in an earlier summary HERE. A reaction is favorable if ΔrG° is negative. ΔrG° comprises two terms, the enthalpy change of reaction (ΔH°), which is related to the stability of the starting materials and products, and the entropy change of reaction (ΔS°). The latter can be simplified to a measure of disorder or randomness.

Relating the change in Gibbs free energy with enthalpy and entropy.
In an elimination reaction, the number of molecules increases. There are normally two starting materials, the substrate and the base while there are three products, the alkene, the protonated base and a leaving group. This means ΔS° is positive, which is good. There is no change in entropy in a substitution reaction. There are two molecules of starting material and two of product.
If you increase the temperature of the reaction, the entropy term becomes more important for an elimination and ΔrG° becomes more negative. Increasing the temperature of a substitution is less important as entropy hasn't changed. This means increasing the temperature makes eliminations more probable but not substitutions.

Increasing the temperature favors elimination (E1 or E2) over substitution. There is no change in the number of molecules on either side of a substitution reaction and this means is the change in entropy (disorder) is minimal. As temperature amplifies this term, changing temperature has little effect on the Gibbs free energy of a substitution reaction. Eliminations are different. The overall number of molecules increases and so disorder increases. The entropy term is positive. Any increase in temperature magnifies this making the change in Gibbs free energy for the reaction more negative or more favorable.
Increasing the temperature favors elimination over substitution.
Conclusion
This summary has looked at two common mechanisms for elimination reactions and the formation of alkenes. The two mechanisms are E1 and E2. These differ in the timing of the lose of the leaving group. An E1 elimination is a first order reaction. The rate determining step is unimolecular and involves ionization of the substrate to give a carbocation and the leaving group. In a second step, a base removes a proton on an α carbon to form an alkene. E2 elimination is second order. It is a concerted process in which the base attacks the proton promoting formation of the π bond by kicking out the leaving group.
E2 eliminations are normally regio- and stereoselective but can be regio- and stereospecific depending on the structure of the substrate. The difference between selective and specific is found in the reaction mechanism. The mechanism of a specific reaction leads to a single isomer while that of a selective reaction can give more than one isomer but often favors one over the other. The selectivity or specificity of E2 eliminations arises as this mechanism only operates when the proton and the leaving group are antiperiplanar to each other. This controls the reactive conformation of the substrate. In terms of regioselectivity, if there is a choice of protons that can be removed, E2 elimination will favor formation of more stable, more substituted product. This is also known as the Zaitsev product. The regioselectivity can be reversed by using a sterically demanding base that preferentially removes a primary proton to give the less substituted alkene (Hofmann product or anti-Zaitsev product).
Similarly, if there is a choice of diastereotopic protons that can be eliminated, E2 elimination is stereoselective for the more stable E or trans-alkene, with the largest substituents on opposite sides of the alkene. If there is no choice, the reaction is stereospecific and will deliver the geometry of alkene determined by the requirement for an antiperiplanar conformation.
In cyclohexane derivatives, the requirement for the proton and leaving group to be antiperiplanar means that E2 eliminations only occur when the leaving group and the appropriate proton are both in axial positions (sometimes called trans diaxial). Again, this means some eliminations will be regiospecific.
E1 eliminations have more freedom with the only conformational requirement being that the proton being removed should be parallel to the carbocation intermediate. E1 elimination is regio- and stereoselective. It favors the more substituted alkene and the E or trans-geometry.

A brief summary of the mechanisms of elimination.
E1 and E2 elimination mirror SN1 and SN2 substitution reactions, with E1 and SN1 sharing a common intermediate, the carbocation, before a second step delivers the product. E2 and SN2 are both concerted, single step reactions. As a result, all the mechanisms are in competition and most reactions will give a mixture of products.
I have given some simplified generalizations that help start to predict which reactions will lead to which products through which pathway. It should always be remembered that all the factors are acting at once and you need to consider them all when trying to predict the outcome. The summary table below hopefully acts as a good starting point.
