Substitution Reactions (on Saturated Carbons)
Introduction
This is the reaction many traditional organic chemistry courses start with, substitution on a saturated carbon atom by either an SN1 or SN2 mechanism. I don't agree with this approach and start my discussion of substitution reactions with acyl substitution of carboxylic acid derivatives (HERE). I think the chemistry of carboxylic acids is far more useful for most students taking a first year chemistry course, but that is a discussion for another day (and over a pint). Acyl substitutions can be described by the following general mechanism:

A simplified mechanism for acyl substitution. It shows that overall a nucleophile replaces a suitable leaving group (the substitution) but that the reaction mechanism proceeds by nucleophilic attack on the carbonyl group and then collapse of the tetrahedral intermediate. This expels the leaving group and gives the product.
In acyl substitution, a nucleophilic reactant displaces a suitable leaving group to give a new carboxylic acid derivative in a minimum of two steps (it is not direct substitution). The nucleophile donates two electrons to create a new bond. It attacks the reactive carbonyl group to give a tetrahedral intermediate. This collapses, regenerating the carbonyl group and eliminating the leaving group. A good leaving group is one that can take the extra electrons away so that the atoms maintain a full octet. A good leaving group will be a weak base or strong conjugate acid. In this example, it will be a stable anion.
In this summary, I will discuss a substitution involving the exchange of a nucleophile and a leaving group, but now there is no carbonyl group and there must be a different mechanism (or two). The good news is that a lot of the principles cover earlier (with carboxylic acid derivatives) are going to be the same.
As is always the case, this is just the introduction to this topic (it is more complicated than we first reveal but you have to start somewhere (it turns out that some reactions taught as SN1 probably don’t proceed through a carbocation but an alkene instead see HERE)). It is meant as a summary that jogs your memory of what you have been taught elsewhere.

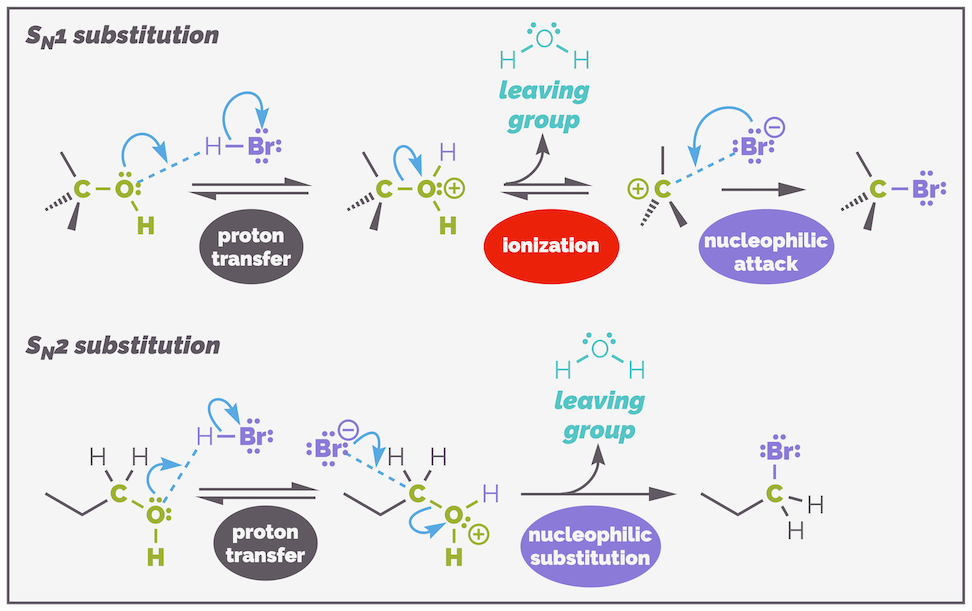
Substitution at a saturated carbon atom.
Substitution Reactions
Below is a classic set of substitution reactions. This is the hydrolysis of bromoalkanes or the substitution of bromide by water. It is a general reaction but I have included the key observations involving the kinetics or rate of the reaction:

Hydrolysis of a bromoalkane is an example of a substitution reaction. Different bromoalkanes react at different rates. What is interesting is that the two structural extremes react the fastest. This suggests that there are two different mechanisms operating.
As you can see, different types of bromoalkane react at different rates. Before discussing the mechanism of the substitution, just a quick mention about the nomenclature of haloalkanes (and all C(sp3)–heteroatom containing compounds).
The carbon bearing a heteroatom (or functional group) can be classified by what other carbon atoms it is attached to. This naming system works as follows: An sp3 carbon is tetrahedral in shape and has four σ bonds. One of these bonds goes to the bromine atom (or heteroatom), any differences come in what the other three are joined to. If there are three hydrogen atoms, the heteroatom is joined to a methyl group. If there are two hydrogen atoms and a single alkyl group, the substrate is classified as primary (1y or 1°), and the heteroatom is said to be on a primary position or primary carbon. If there is only one hydrogen and two alkyl groups it is a secondary (2y or 2°) substrate or the heteroatom is on a secondary position or secondary carbon. If there are no hydrogen atoms and only alkyl groups, tertiary (3y or 3°), and the heteroatom is said to be on a tertiary position or tertiary carbon. This classification can get confusing when discussing amines. Amines are classified as primary, secondary, and tertiary depending on how many alkyl groups are bonded to the nitrogen, but where they are situated on the alkyl chain is also categorized as primary, secondary and tertiary by the pattern given above. Annoyingly, the three words are being used to mean two different things and this means a substrate can be both a primary and a secondary amine (for example) depending on whether you are discussing the nitrogen of the amine or the carbon that nitrogen is attached to … whoever invented the naming system hates you!

Substrates can be classified depending on the number of alkyl groups attached to the carbon that is bonded to a functional group or heteroatom.
The reason that both bromomethane and the very bulky tert-butyl bromide (2-bromo-2-methylpropane) react quicker than secondary bromides, even though they are structurally very different is that there are two substitution mechanisms operating (there are more, but that is a story for another day). One mechanism favors accessible leaving groups such as those found on methyl and primary substrates while the other favors tertiary (and bulky) substrates. As secondary positions sit in the middle, they aren’t particularly good for either mechanism.
The mechanisms are called SN1 and SN2. The S stands for substitution, the subscript N means nucleophilic as the substrate is always attacked by a nucleophile. The two mechanisms differ (obviously) by the number, and this refers to the kinetics of the reaction. SN1 has a single molecule in the rate determining step (RDS), it is first order, while SN2 has two reactants in the RDS or is second order (bimolecular).
Below, I’ll go into more details about the two mechanisms, their differences, and how you can start to predict which substrates will react through each mechanism. When first learning mechanisms, it is easy to start with these extremes, but I think it is important to realize that most reactions lie somewhere in between, and are not 100% concerted nor necessarily 100% stepwise (it’s confusing but hopefully this summary will help!).
SN2 Mechanism
This is a second order reaction with both substrates, the nucleophile and the electrophile, in the rate determining step RDS. The mechanism involves the two reactants coming together in a single step. There are two curly arrows, one to show the formation of the new carbon–nucleophile bond and one to show the breaking of the carbon-leaving group bond. The reaction is concerted with the new bond forming at the same time as the old bond breaks.

The mechanism of an SN2 substitution involves the concerted formation of the new C–nuc bond and breaking of the C–LG bond. There are two molecules in the rate determining step. This is a single elementary step, meaning the mechanism cannot be broken down into any other reaction and there is no intermediate. This is represented by two curly arrows.
The reaction profile for the transformation is shown below. It shows that the reaction is concerted, being comprised of a single elementary step (if you believe in that kind of thing) with a single transition state. In the transition state there are two partial bonds with the charge spread across them both (the carbon does not have five bonds. These are partial bonds starting to form and starting to break. They are effectively half bonds).

A reaction profile for a typical SN2 substitution showing the transition state with partial bonds between carbon and both the nucleophile and leaving group.
The transition state reveals an important characteristic of SN2 substitution, one that differentiates this reaction from SN1 substitution. The reaction occurs with inversion of stereochemistry (if the leaving group is on a carbon with three different substituents in addition to the leaving group). The nucleophile must approach from 180° to the leaving group, in what is often termed backside attack. This leads to the substituents of the stereocentre being pushed backwards, rather like an umbrella being blown inside out in the wind. This inversion of stereochemistry is sometimes given the name Walden inversion.

SN2 substitution occurs with inversion of stereochemistry (should the electrophilic carbon be a stereocenter).
The transition state shows a partial bond between the nucleophile, in this case the sulfur atom, and carbon along with a partial bond as the leaving group, a bromide here, starts to depart. The carbon does not have five bonds. The dashed lines represent the start of a bond forming and the start of a bond breaking. The charge of the incoming anion is no longer localized on a single atom but is spread across three as the extra electrons brought by the nucleophile are taken away by the leaving group. This is indicated by the partial charges on the incoming and departing groups. The fact that these aren't full bonds is why I haven't drawn curly arrows on this version of the reaction
The transition state gives you a clue as to the properties needed in a good leaving group. The leaving group must polarize the bond, making the carbon electron poor so that the nucleophile will attack. The leaving group also needs to be capable of taking two electrons away from the substrate. In other words it needs to form a stable or weak base. I’ll go into the properties of the leaving group in more detail when discussing how to identify the two mechanisms in the wild.
A quick inspection of the orbitals involved in SN2 substitution reveal why the nucleophile approaches from 180° to the leaving group. Using the simple frontier orbital approach (based on valence bond theory) favored by organic chemists, the reaction involves the highest occupied molecular orbital (HOMO) of the nucleophile overlapping with the lowest unoccupied molecular orbital (LUMO) of the electrophile. In the example above, HOMO is a lone pair of electrons (it is always a full orbital regardless of the nucleophile) while the LUMO is the empty σ* antibonding orbital of the carbon-leaving group bond. The largest coefficient of the this antibonding orbital is 180° to the bonding orbital.

The frontier molecular orbital approximation of an SN2 substitution. It shows the HOMO of the nucleophile overlap with the LUMO of the electrophile, which leads to the formation of a new σ bond and the breaking of an old σ bond. Bonding forming and breaking passes through a transition state in which the carbon atom is approximately trigonal planar and the electrons of the nucleophile, substrate and leaving group are shared through a p orbital. The only way this achievable is if the incoming nucleophile and the outgoing leaving group are on opposite sides or are 180° to each other.
The carbon atom of the transition state is effectively trigonal planar with a p orbital running through the center. This p orbital shares a pair of electrons with the incoming nucleophile and a pair with the departing leaving group. It shows that the two must be at 180° to each other and this starts to explain the necessity of backside attack.
SN1 Mechanism
At the other extreme is the SN1 mechanism. This is a first order reaction with the rate of reaction being controlled by the substrate only. This means there is only one molecule in the rate determining step; the nucleophile has nothing to do with the rate of reaction. As you can see from the reaction mechanism below, the SN1 substitution is a stepwise process that proceeds through the formation of a carbocation intermediate. The mechanism involves two discreet steps, both have only a single curly arrow. The first step, the ionization (or dissociation or elimination or explusion) is rate determining.

The mechanism of the second substitution reaction. This is SN1 substitution and is a stepwise process that involves the elimination of the leaving group prior to the nucleophile attacking. The first step is the rate determining step.
In the first step of the reaction the C–LG bond breaks and the leaving group dissociates (sometimes it is said to be eliminated, or expelled, or ionized … bottomline, it leaves but I don’t like writing ‘leaving group leaves’, it makes for a clunky sentence). This is the rate determining step as it has a large activation energy as represented on the energy profile below. This makes sense, you are breaking a bond, which takes energy, and there is no compensation until the nucleophile attacks and a new bond is formed. The second step leading to the new C–nucleophile bond is fast as it releases energy.

The SN1 substitution is a stepwise process that involves elimination and then addition. The first step, the lose of the leaving group (elimination) is the slow, rate rate determining step due to the size of the activation energy. Addition of the nucleophile is rapid.
Key to understanding SN1 (at undergraduate ... just to keep some of my more pedantic colleagues happy) is the intermediate carbocation. The more stable this intermediate, the faster the reaction will be, and the more likely the reaction is to proceed by an SN1 mechanism. The carbocation also explains why this substitution can lead to two products. When the leaving group is lost, the tetrahedral sp3 hybridized carbon atom becomes a trigonal planar sp2 hybridized. In the second step, the nucleophile can approach from either side leading to two different enantiomers (if the starting material was chiral).
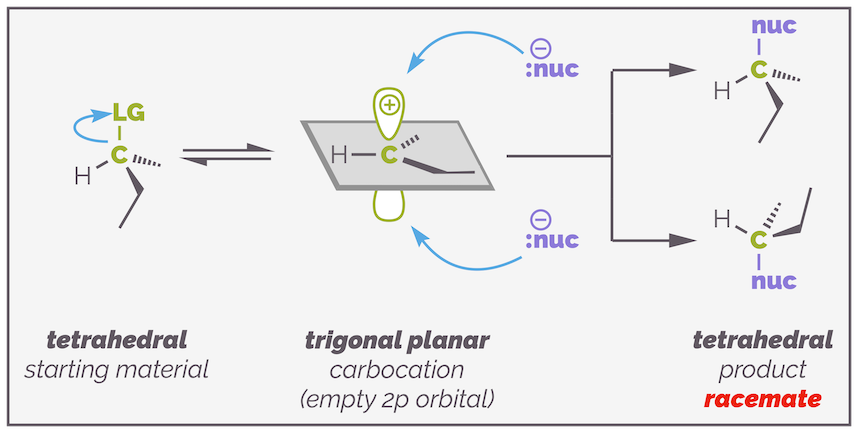
When the substrate is chiral and the leaving group is on the stereocenter, the SN1 mechanism can give rise to a racemate. Once the leaving group departs, you are left with a trigonal planar carbocation. This flat intermediate can be attacked from either side leading to a mixture of enantiomers. This diagram tries to show the flat intermediate by placing all the substituents in the dark grey plane.
As we shall see later, secondary substrates are problematic. They can potentially proceed by either SN1 or SN2. There seems to be some discussion in the literature as to what is actually happening (HERE).
While this description of the scrambling of stereochemical information is found in all textbooks, it isn't necessarily true. I'm going to ignore molecules with multiple stereocenters and the fact that the shape of the rest of the molecule can influence which face of the carbocation is attacked (or substrate controlled reactions), and just deal with the example above with its single stereocenter. It is rare to observe complete racemization in the SN1 reaction. Ionization occurs to give a pair of ions, the carbocation and the anion. As opposite charges attract, the leaving group doesn't completely drift off. Instead, a tight or intimate ion pair is formed. The leaving group, with its bulk and negative charge hampers the approach of the electron rich nucleophile. This means the nucleophile predominantly approaches opposite the leaving group and a greater proportion of inversion of configuration is observed.

Chemistry is rarely as simple as your lecturer will make out. This even includes SN1, one of the first reactions covered in many courses (but not my own I hasten to add, and will argue why it should be at long length given half the chance), is more tricky than made out. It is always stated that the reaction occurs with lose of stereochemical information and formation of a racemate if a chiral substrate was used. This isn't always true. Tight (intimate) ion pairs form, meaning that the nucleophile attacks before the leaving group has completely departed. This means that you normally observe a little more inversion than might be expected.
SN1 versus SN2
Hopefully it is apparent that the difference between the two substitution mechanisms is one of timing or when the leaving group departs (when it leaves). For SN1, the leaving group is expelled before the nucleophile attacks while in SN2 the events are simultaneous. This means the big difference comes down to the formation, or not, of the carbocation intermediate. The more stable the intermediate the more chance the reaction is SN1.

The two substitution mechanisms discussed in this summary (there are more), differ by the timing of the leaving group departing from the substrate. It either occurs before the nucleophile attacks or as the nucleophile attacks. This means the difference between the two mechanisms often comes down to the stability of the carbocationic intermediate.
While this simplification is useful, please be aware that most substitution reactions occupy a grey area in between these two extremes. This was alluded to when I mentioned the existence of the tight or intimate ion pairs.
Predicting SN1 versus SN2
Often, for undergraduates, the hardest part of learning about substitution mechanisms is trying to use all the information you are given to predict which substrates and which reactions proceed through which mechanism. It is hard, and it is often not clear (the number of textbooks that state tertiary leaving groups never participate in SN2 substitution is scary (becuase it is wrong as you shall see)).
While all factors of a reaction influence the mechanism, and many of the factors influence each other (solvent is more important than textbooks (and I) give it credit for), I'm going to simplify the discussion and concentrate on four (or three depending on how you want to count these). You’ve got to start somewhere! These are:
Substrate structure
The leaving group
The nucleophile
The solvent
The first two concern the structure of the electrophile so could count as the same factor (but hopefully, you will see why I have split them up). As the substrate or electrophile appears in the rate law for both mechanisms it is very important. The solvent is equally important as virtually all reactions are conducted in a solvent but, if you look at enough textbooks, you will notice that it is invariably ignored (not even written next to the reaction arrow). This is bad practice but I’m happily going to follow in the footsteps of my published brethren as this summary is already too long …
1. Substrate structure (ignoring leaving group)
If the substrate cannot form a relatively stable carbocation then it will not react through an SN1 mechanism. Or, put another way, the more stable the carbocation the more probable SN1 is. What factors influence cation stability? There are two you need to think about. This first is the inductive effect, hyperconjugation or sigma bond delocalization (three names for the same effect).
If you ignore resonance, then the stability of carbocations follows this order: tertiary carbocations are more stable than secondary cations, which are more stable than primary cations, and methyl cations are the least stable. Alkyl groups are electron donating and the more electron donating groups that are attached to an electron deficient carbocation the more stable it is.

The stability of simple carbocations is determined by hyperconjugation or the inductive effect (two names for the same concept).
One way to understand why alkyl groups are electron donating looks at the orbitals (or at least valance bond overview; for most first year courses, this is more information than you require). A carbocation is a carbon with only six valence electrons. It has an empty 2p orbital and a positive charge. Hyperconjugation, or σ donation, describes the sharing of electrons from adjacent C-H or C-C σ bonding orbitals into this empty 2p orbital. It can be thought of as σ bond delocalisation. Effective sharing can only occur if the orbitals are parallel allowing full overlap. Only one bonding orbital per alkyl group can align. Thus more alkyl groups on the carbocation the more hyperconjugation and hence greater stability.

Hyperconjugation is the delocalization of σ electrons by the overlap of σ bonds.
This all supposes that the carbocation can adopt a trigonal planar carbon atom. Some cyclic structures (especially those with bridges) prevent the carbocation from being trigonal planar and reduce the degree of hyperconjugation. This means the carbocation is less stable. The shape of molecules is very important.
According to the stability of the carbocation intermediate, tertiary 3° substrates favor SN1 while primary 1° (or methyl) react through SN2.
I've fallen into the same trap most textbooks do when discussing SN1, and have stated that the reactions occur because of the stability of the carbocation. This is not strictly true. Whether a substrate reacts by SN1 or SN2 is determined by which mechanism is faster. This in turn, if you remember our discussion of rates of reaction, is controlled by the activation energy or the energy of the transition state (lower energy transition state, faster reaction). So what I should be typing is that tertiary electrophiles lead to a transition state with lower energy. What does the transition state look like? The Hammond postulate tells you that it resembles whichever stable species (adjacent in the reaction profile) it is closest in energy to. This means you look at the reaction profile above, it means the transition state of SN1 looks like a cation ... and hence all our arguments are more or less valid. Yay! (I've fallen into a number of other gross simplifications but I'm sure you will forgive me (while some of my colleagues will not)).
Delocalization of electrons will also stabilize a carbocation, and this can promote SN1 reactions but, as you shall see, delocalization will also increases the rate of SN2 reactions. Benzyl chloride is a primary halide. Normally, this would indicate that the reaction should be SN2 but the carbocation is delocalized over the benzene ring. This encourages SN1. Ultimately, it can react by either mechanism and other factors will determine the mechanism.
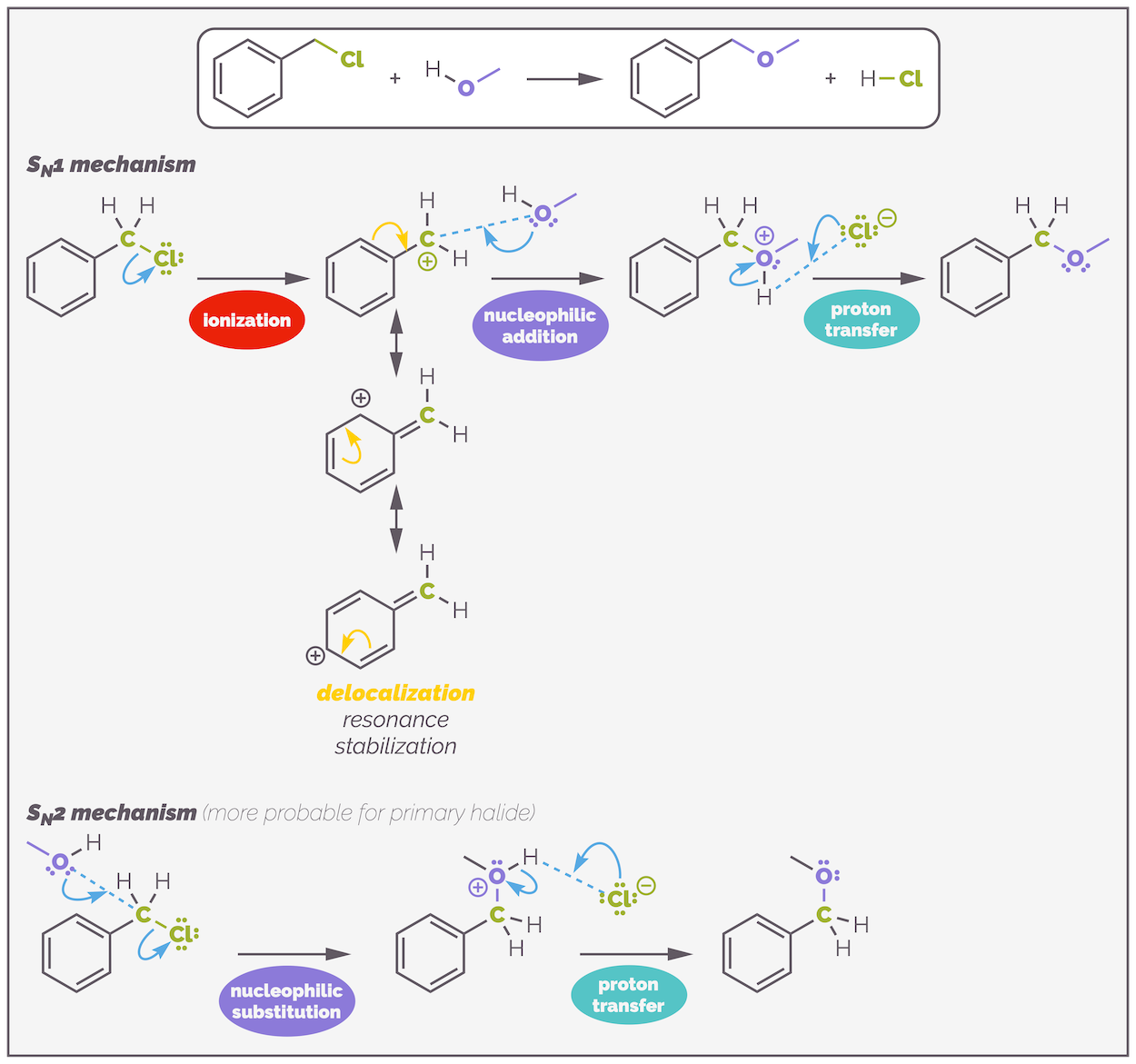
Substitution of a halide on a benzylic position can be either SN1 or SN2. Both mechanisms are enhanced through conjugation. A primary halide, such as the one in this example, is probably SN2 due to the rapid attack on the primary position (although, I have muddied the waters by using a poor nucleophile). If the halide had been either secondary or tertiary then more SN1 would be observed. In these two examples, there is a deprotonation step to give the neutral final product. This step is rapid and makes no difference to the rate of reaction or mechanism. Almost certainly, the base would actually be the alcohol and not the chloride shown but I've used the chloride to balance the equation (this is one of the many ways chemistry tries to confuse students). Solvent will play a role in determining mechanism but that is a subject for another day.
Conjugation or delocalization also enhances the rate of SN2 substitution. In the transition state, there are two partial bonds to the carbon atom. Effectively, this means two electrons are being shared across three atoms, and there is a build up of positive charge on the carbon. Remember our description of the transition state above, it stated that you can think of the carbon sharing an empty 2p orbital with the incoming nucleophile and the outgoing leaving group. That empty orbital represents the build up of positive charge. Delocalization of the adjacent π electrons stabilizes the partial positive charge (or the less than full 2p orbital on carbon). This in turn leads to lowering the energy of the transition state or lowering the activation energy and the rate of reaction increases.

Conjugation stabilizes the transition state of SN2 substitution. The π electrons are delocalized onto the partially positive carbon. This lowers the energy of the transition state and the rate of reaction increases. This means delocalization can aid both reaction mechanisms.
The stability of the carbocation is not the only way the structure of the substrate can influence the reaction mechanism. For a reaction to proceed by the SN2 mechanism the nucleophile should be able to approach the substrate from 180° to the leaving group. This is easy for a methyl group or a primary leaving group as there is little steric hindrance blocking attack, but as more substituents are added the reaction becomes slower. Secondary leaving groups react slower than primary examples while tertiary groups are virtually inert to SN2. Hopefully, this is shown in the diagram below:

For an SN2 reaction the nucleophile must approach from 180° to the leaving group. This is easy for methyl and primary substrates but when the leaving group is on a tertiary position there is so much steric bulk that the nucleophile finds it hard to approach.
The steric bulk that results from multiple substituents on the reactive carbon favors the SN1 mechanism for a second reason. In the starting material, the leaving group is on a tetrahedral sp3 hybridized carbon atom. The bond angles are 109°, and this forces the substituents relatively close to each other. Reaction through an SN2 mechanisms leads to a transition state that resembles a five coordinate species. The three substituents are 120° to each other but they are all 90° to the nucleophile and the leaving group. That is six disfavored interactions as the groups are now closer. The increase in strain disfavors or slows SN2 by making the transition state, and the activation energy, far higher.

The substituents on the electrophilic carbon atom can influence the reaction in multiple ways. One factor that is often over looked is the change in bond angle. SN2 is disfavored if there are multiple substituents as the transition state involves bringing multiple groups closer together while bulky groups accelerate the SN1 mechanism as the transition state of the rate determining step allows groups to move apart.
If the reaction proceeds by the SN1 mechanism, elimination of the leaving group forms a trigonal planar carbocation (it is sp2 hybridized with an empty 2p orbital). The bond angles are 120° and the substituents are further apart than in the starting material. The transition state for the ionization step is favorable as it is leading to an increase in the bond angle and a release of strain in tertiary compounds. Overall, this speeds up or encourages SN1 reactions.
The more observant will have notice that the structural factors that promote SN1 (tertiary better than primary) are the opposite to those that favor SN2 (primary better than tertiary). This is good as they reinforce each other. It means the majority of tertiary substrates will react by SN1 while the majority of primary substrates react by SN2 (all other factors being equal). Secondary molecules are more problematic being poor substrates for both mechanisms.
Simply looking at the structure of the electrophile, a number of generalizations can be made:
- Tertiary electrophiles favor SN1 and are bad substrates for SN2
- Primary (& methyl) electrophiles favor SN2 and are bad substrates for SN1
- Secondary electrophiles are moderately capable of either mechanism
- Conjugation or delocalization enhances both mechanisms by either stabilizing the cationic intermediate (SN1) or by stabilizing the build up of positive charge on the carbon in the transition state of an SN2 substitution.
I've been a bit naughty and left one example to the end so that we can discuss how these various factors influence the mechanism of substitution. Below is the substitution of a tertiary bromide. Would the reaction proceed by an SN1 or SN2 mechanism?
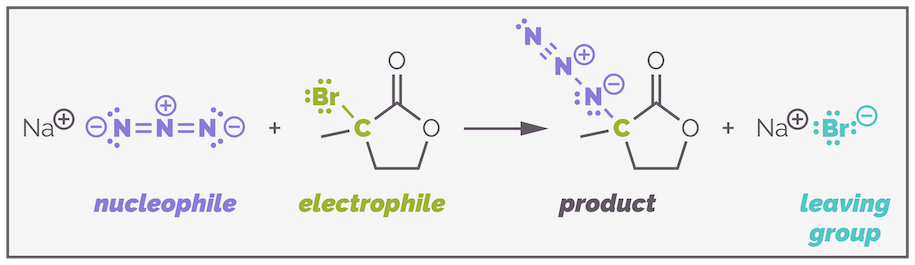
An example of a challenging substitution. Which mechanism is operating?
Many students look at this example and instantly think it must be SN1. It is so common to read that tertiary positions will not react through an SN2 mechanism due to sterics, but this statement simply isn't true. There is invariably a competition between the two mechanisms and you observe the faster reaction. It is necessary to determine which mechanism is more likely out of two by assessing all the possible factors.
It is obvious why SN2 might be disfavored (steric hindrance) but what is the problem with the SN1 mechanism? SN1 is favored when the carbocation is stabilized. Here, the carbocation is α to a carbonyl group that will destabilize the cation. Electron withdrawing groups disfavor the build up of a positive charge; you are effectively adding a cation next to a highly polarized partially positive carbon of the carbonyl. This is not good.

SN1 substitution next to a carbonyl group is disfavored as the formation of a carbocation next to a highly polarised carbon with a partial positive charge is energetically unfavorable (the transition state will be high energy).
SN2 next to a carbonyl group is a fast process. In fact, it is faster than 'normal' SN2 processes. When the leaving group is on the adjacent carbon to the carbonyl group, the so-called α-position, the two electrophilic groups can interact. There is an overlap, or combination, of the two antibonding orbitals. The carbonyl π antibonding orbital and the leaving group's σ antibonding orbital mix to create a new, lower energy LUMO. The electrophile is now even more electrophilic and substitution occurs more rapidly. This acceleration overcomes the issue of sterics.

A halide adjacent to a carbonyl group is around reacts ~100,000 times faster than the analogous primary halide. The reason for this is the conjugation of the antibonding orbitals leads to a lower energy LUMO. This reacts faster with nucleophiles and can offset other issues ...
In the case of the original question, SN1 is disfavored due to presence of the electron withdrawing ester. SN2 is disfavored due to the steric hindrance caused by a tertiary position but is accelerated by the α-carbonyl group. Overall, SN1 is very slow while SN2 isn't fast but it is more favorable.

For this question, the answer is that the reaction proceeds by an SN2 mechanism. The reaction will be slow, as the steric hindrance of a tertiary position will slow the reaction considerably, but it is still more favorable to have SN2 than SN1 as there is no cationic intermediate and α-halides react by SN2 faster than normal.
So here are some generalizations about how substrate structure influences mechanism (these are just generalizations and there are many examples that don't appear to follow these but do follow sound chemical pronciples that will be covered at a later stage. In other words, these are a good starting point and unless you are in your third year or your instructor hates you, they should be good):
- Substrates that can stabilize a carbocation favor SN1 substitution.
- Bulky or sterically hindered substrates favor SN1 substitution.
- Primary substrates favor SN2.
- Methyl groups must react by SN2.
2. Substrate structure - the leaving group
The nature of the leaving group influences the rate of both reactions as the substrate is involved in the rate determining step of both mechanisms or is found in the rate equation for both. The better the leaving group the faster the rate of both reactions. I don’t want to sound like a stuck record but this is a summary that covers the basics, as always, the topic is more complex (leaving group ability is influenced by many factors not discussed here especially solvation) … trust me, the subject is both fascinating and confusing but rarely do students need to go into detail.

Rate equations showing that the leaving groups is involved in the rate determining step of both substitution mechanisms.
It turns out that the SN1 mechanism is more sensitive to the nature of the leaving group as the rate is solely controlled by ionization of the substrate. The rate of SN2 reactions is controlled by both substrate and the attack of the nucleophile. The effect of the substrate on SN2 is somewhat diluted (pun intended).
The rate of both mechanisms increases if the substrate has a good leaving group. When a good leaving group is kicked out of the substrate it forms a weak base (or the species has a strong conjugate acid with a low pKaH). This means the more stable an anion, the better the leaving group while neutral groups are better than anionic etc.

Good leaving groups form weak bases when they are eliminated from the substrate. Iodides are better leaving groups than fluorides (the C–F bond is the strongest single bond in common organic compounds and normally only behaves as a leaving group in nucleophilic aromatic substitution). Elimination of a neutral species will be better than formation of an anionic species.
Iodide is a better leaving group than fluoride. It is less basic, forming a more stable anion due to the charge being spread over a large ion. This is demonstrated if you compare the pKaH of HI and HF (–10 and 3.2). In addition, the C–F bond is one of the strongest single bonds (450 kJ mol-1) especially when compared to the weak C–I bond (240 kJ mol-1). (as an aside: Fluoride can appear to be a good leaving group in nucleophilic aromatic substitution but it enhances this reaction for very different reasons and is still effectively a poor leaving group (see HERE)).
Alcohols are a common functional group found in many molecules but they are not (directly) useful in substitution reactions. The hydroxide anion (HO-) is a poor leaving group. It has a pKaH = 14 and can be considered a strong base. Hydroxide is never a leaving group in SN2 reactions. It is a rubbish leaving group, and if you use a nucleophile that is sufficiently strong nucleophile to kick it out, it still wouldn't as the nucleophile will react with the proton of the alcohol faster, leading to deprotonation and the formation of an alkoxide.
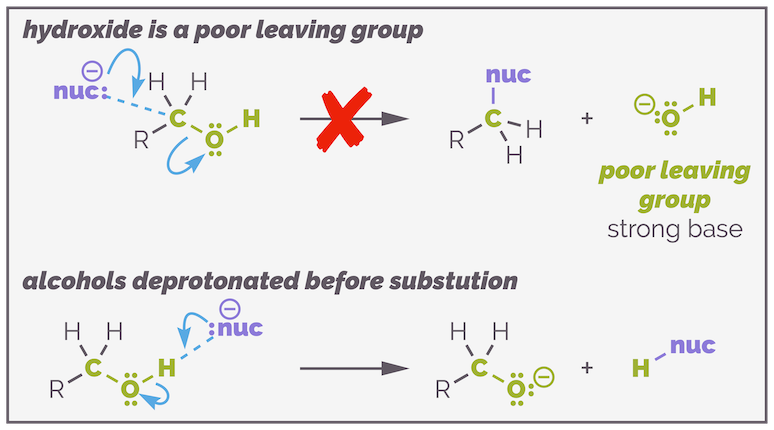
The hydroxide anion is never a leaving group in SN1 and SN2 reactions. It is a strong base so is a poor leaving group. Also the proton of an alcohol is relatively acidic and will be attacked by any nucleophile sufficiently strong to attempt substitution.
To substituted an alcohol, it must first be converted into a good leaving group. The easiest way to achieve this is by reaction with a strong acid. This protonates the alcohol to give an oxonium ion, and now water is the leaving group. Water (H2O) is a good leaving group, with a pKaH = 0. This allows the formation of alkyl halides by either SN1 or SN2 mechanisms. In the SN1 reaction, the oxonium will depart to give a carbocation while in SN2 reactions it is directly displaced.
Alcohols can be converted into good leaving groups by treating with acid. The resulting protonated oxonium ion is a good leaving group and will participate in both SN1 and SN2 reactions depending on the substrate structure.
Another way to make alcohols into good leaving groups is to convert them into sulfonate esters such as mesylates (R–OMs), tosylates (R–OTs) or triflates (R–OTf). The first two are easily prepared by reaction with appropriate chlorides, either methanesulfonyl chloride (mesyl chloride, MsCl) or p-toluenesulfonyl chloride (tosyl chloride or TsCl). Triflates are more reactive and they are formed from the anhydride.

The synthesis of the common sulfonate esters. The products are shown in skeletal representation and abbreviated, using the organic elements Ms, Ts, and Tf to represent mesylates, tosylates and triflates.
Substitution of a sulfonate ester results in a sulfonate anion (RSO3–) being expelled as the leaving group. The pKa of the conjugate acids indicate that these three sulfonates are good leaving groups; MsOH = –1.9, TsOH = –2.8, and TfOH = –14.7. The anion is stabilized by the electronegativity of the oxygen atoms and delocalization of the charge. The triflate is the most reactive as a result of the increased stability resulting from the inductive effect of the three fluorine atoms. Each of the sulfonates can be substituted by most nucleophiles.
Having formed a good leaving group, the chemistry is the same as discussed above and reactions can occur by either SN1 or SN2 mechanisms. Which mechanism is in operation will be controlled by other factors (remember, as the leaving group is present in both rate determining steps, making a better leaving group enhances both mechanisms and isn't good at differentiating the two). The first example is SN1 as the substrate contains both a tertiary leaving group and it next to an aromatic ring. This means it is easy to form a stabilized carbocation and this favours SN1. It should also be noted that the use of a weak nucleophile (a thiol) also encourages an SN1 mechanism, as will be discussed in the next section. The scrambling of the stereochemistry, going from a single enantiomer to a mixture of enantiomers indicates that this is a stepwise process proceeding through achiral intermediate.

The substitution of sulfonate esters. The top shows the substitution of a tosylate while the bottom shows the substitution of a mesylate. The top reaction proceeds through an SN1 mechanism while the bottom is SN2; this change in mechanism is not due to altering the sulfonate ester but rather changes in the substrate structure.
The second example shows the substitution of a mesylate by an SN2 mechanism. In this example, the carbocation is not that stable and the use of a strong nucleophile, which actively attacks the substrate, both result in SN2 is favoured. The fact that the substitution occurs with inversion of stereochemistry, maintaining a single enantiomer, tells you that a carbocation was not formed.
- A good leaving group accelerates both SN1 and SN2.
- HO– is never a leaving group in either SN1 and SN2 reactions.
3. Nucleophile
The structure of the nucleophile can influence the mechanism of substitution. The nucleophile appears in the rate equation for SN2 reactions. This means the rate of an SN2 reaction does depend on the strength of the nucleophile. A good or strong nucleophile will speed up an SN2 reaction while a a poor or weak nucleophile will not. The structure of the nucleophile has no effect on the rate of an SN1 reaction; it does not appear in the rate equation.
Strong nucleophiles encourage SN2 reactions as they attack the substrate. Weak nucleophiles disfavor SN2 reactions as they don't attack the substrate. This allows SN1 reactions to occur and they become a competing pathway. So, while the nucleophile doesn't actively encourage SN1 reactions it can cause a swap from one mechanistic pathway to the other.
But what is a strong nucleophile? Good nucleophiles readily donate a pair of electrons to create a new bond. In a different summary (HERE), I discussed how to predict the relative basicity of various compounds. Simplistically, bases are nucleophiles that only attack protons (nucleophiles form bonds to carbon while bases form bonds to hydrogen). This means you can use basicity as a starting point for predicting relative nucleophilicity or nucleophile strength. A good or strong nucleophile will often be a strong base and this means it will have a weak conjugate acid (a conjugate acid with a high pKaH). But you must be aware that this is on a starting point. Firstly, comparing pKaH is only good in certain specific cases. Other factors, such as solvation, are important. Secondly, while steric hindrance rarely influences basicity, as you are looking at the attack on a very small species, a proton, it plays a major role in nucleophilicity, where it can prevent reactants from interacting.
When looking at the relative strength of nucleophiles, the first factor to assess is charge. It is often stated that anions are more nucleophilic than neutral species, and this is true as long as the structures are similar, but please remember that you must assess all factors. The conjugate base of a species is always the stronger nucleophile.

Anions are invariably better nucleophiles than neutral species (although it would be better to write that the conjugate base is always the stronger nucleophile so that you don't start comparing an anion of one species with a the neutral form of a completely different molecule). When comparing nucleophilicity, you can use pKa as a starting point, remembering that a good nucleophile is often a good base and so you are looking for the species with the high pKa for the conjugate acid. But two things that trip the unwary, first, make sure you are comparing the conjugate acids, and secondly, pKa is not a guide if the nucleophilic atoms are in different rows so be careful.
Another important factor is electronegativity BUT only when you are comparing atoms in the same row of the periodic table. As you move along a row the electronegativity increases and the atom holds onto its electrons more tightly. It is less likely to donate them and is a weaker nucleophile. This is true comparing anions or neutral species.

As you move across a row of the periodic table the nucleophilicity decreases as the electronegativity increases. This effectively matches the ease of donation of a lone pair of electrons. More electronegative elements hold on to the lone pair more and so it is harder for it to be shared; the species is less nucleophilic. This allows you to use pKa as a predictor, but only as you move across a row.
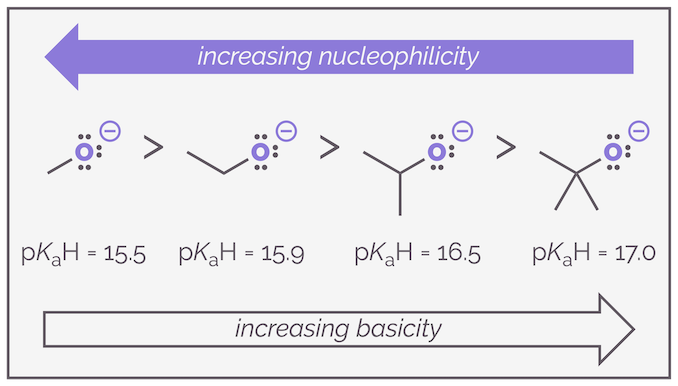
Because of this trend across a row, it is possible to use pKaH values to estimate nucleophilicity. The higher the pKa of the conjugate acid the greater the chance of the molecule being a good nucleophile. This is shown in the figure above. The use of pKaH values also works quite well if you are comparing the nucleophilicity of the same atom in different molecules. Below is a series of oxygen anions and the nucleophilicity more or less follows the basicity.

Basicity (and acidity) can act as a predictor for relative nucleophilicity but should not be relied upon. It works across a row of the periodic table and with the same atoms ... most of the time but it should always be remembered that acidity is a thermodynamic effect (measured by an equilibrium constant) while nucleophilicity is a rate effect (how fast a reagent attacks another).
But, and I keep stressing this, pKaH values only give an estimate, steric hindrance has a large influence on nucleophilicity, with larger, more hindered molecules being weaker nucleophiles. Sterics has minimal influence on the basicity, and hence pKaH values, of the same compounds.
Steric hindrance influences nucleophilicity but has little effect on basicity. Less hindered compounds are better nucleophiles as they can attack the carbon more readily.
Basicity is not important when moving down a group of the periodic table. Here you should be more concerned with polarizability. Bigger atoms have more electrons and the distance of the outer electron shells from the nucleus increases. This means the electrons of a large atom are more readily polarized or more readily become unevenly distributed. The result is that they are more readily donated to the appropriate electrophile. The atom is more nucleophilic. Bigger atoms are more nucleophilic. This can also be explained by electronegativity, the attraction of the nucleus on the outer electrons decreases as the atom increases in size. The increased nucleophilicity has also been explained by better orbital overlap; as the size of the atom increases so the lone pairs have higher energy (the HOMO is raised) and they are closer in energy to the LUMO. All these explanations explain why the iodide anion is a good nucleophile and the fluoride anion isn’t (ignoring all other factors for the time being).

When comparing the nucleophilicity of atoms in the same group, size or polarizability is more important the basicity. The larger the atom, the more electrons and the polarizable it is. There are more electrons and they are further from the nucleus so they are readily unevenly distributed and more readily donated to an electrophile. With increasing size of an atom the HOMO is raised and becomes closer to the LUMO of the electrophile; the reaction is easier.
Below is a rough guide to strength of nucleophiles (ignoring the effect of solvent for the time being):

Summary of relative nucleophile strengths (ignoring solvation).
Key points:
- A strong nucleophile will favor the SN2 mechanism
- A weak nucleophile disfavors the SN2 mechanism (so more SN1 is observed but the nucleophile never encourages SN1 as it is not in the rate equation).
4. Solvent
To the annoyance of generations of students, the nature of the solvent can influence the mechanism of substitution reactions. Below is only a simplistic overview of the effect of solvent, the kind needed for most undergraduate courses. Many chemists will think I’m doing a disservice to the importance of solvent.
Common solvents are divided into two categories, polar and non-polar solvents. Polar solvents have a large dipole moment, meaning an uneven distribution of electrons and you could draw partial charges (δ+ and δ–) on the structures. They will contain atoms with very different electronegativities. Non-polar solvents are the opposite (surprise), they have bonds between atoms with little different in electronegativity (C and H) and, as a result, have no partial charges. But you need to remember polarity is scale with some molecules being more polar than others. This means there is a lovely grey area in the middle where molecules have both polar and non-polar properties. The polarity of a solvent can be determined by measuring either the dielectric constant or the dipole moment.

Solvents can be divided into two categories, non-polar and polar solvents, although it should always be remembered that this a scale with some solvents being more polar than others (a rough rule of thumb involves water solubility; it if dissolves in water it is polar, if it doesn't it isn't, but this is very crude). Polar solvents can be further subdivided into protic (hydrogen bond donors) and aprotic (not hydrogen bond donors).
Non-polar solvents have small dielectric constants, normally <5 while polar are normally over 20.

Examples of polar and non-polar solvents.
A number of common solvents are in the grey area between polar and non-polar. They have moderately large dielectric constants between 5 and 20. Probably the three most important are shown below:

Examples of borderline polar/non-polar solvents.
Polar solvents, those with a dielectric constant >20, can be further subdivided as either protic or aprotic. Polar protic solvents posses either and O–H or N–H bond and will be both hydrogen bond donors and acceptors. The ability to act as hydrogen bond donors will have a major influence on reactivity. Examples are given below:

Examples of polar protic solvents
Polar aprotic solvents have a large dipole and dielectric constant but cannot be hydrogen bond donors (but they will be hydrogen bond acceptors).

Examples of polar aprotic solvents.
Having categorized solvents as non-polar, polar protic and polar aprotic, how does this influence the mechanism of substitution? In an SN1 substitution, ionization leads to the formation of a carbocation and an anion (it's kind of in the name). Polar solvents will stabilize the carbocation intermediate. This is like dissolving salt in a solvent. You start with a neutral species, NaCl, and you want to solvate both the cation and the anion. This is unlikely to occur in a non-polar solvent as there are no non-covalent interactions between the ions and the solvent. Use a polar solvent and the salt dissolves due to ion-dipole interactions. Stabilizing the intermediate of a reaction results to a lowering in energy of the transition state that leads to the intermediate. This means the activation barrier is lower and the rate of reaction will increase. Polar solvents will increase the rate of SN1 reaction.

Polar solvents stabilize the cationic intermediate. This means it is easier to form the intermediate and the activation energy is lower. Substitutions occurring by an SN1 will be faster in polar solvents.
With SN2 substitution reactions, the nucleophile is important as it is part of the rate equation. The nucleophile tends to be strong and is often anionic. It needs to be as the electrophile in SN2 reactions is weaker than those in SN1 reactions, it's not a carbocation! Anionic nucleophiles, such as NaOH, are salts and only dissolve in polar solvents, and most SN2 substitutions occur in polar solvents. But, polar aprotic solvents are favored rather than protic solvents. Protic solvents stabilize the nucleophile making it less reactive. They form a solvent shell around the nucleophile making it hard for the nucleophile to approach the substrate (solvation). Polar aprotic solvents are favored for SN2 reactions as they do not interact with nucleophile well, leaving it naked and ready to react with the electrophile. Best solvents are often acetone or DMSO.

Polar solvents are normally required to dissolve both the substrate and the nucleophile, with the latter frequently being an anion with a metal counter cation. Polar protic solvents disfavor SN2 substitution as they can hydrogen bond to the anion and create a solvent shell that hinders the approach of the nucleophile and electrophile. Polar aprotic solvents generally favor SN2 substitutions. They are capable of dissolving the reactants but form only weak interactions with nucleophile. They are said to leave the nucleophile naked, which makes it highly reactive and capable of easily attacking the electrophile.
Key points:
- Polar protic solvents favor SN1.
- Polar aprotic solvents favor SN2.
Examples
I'll finish with a number of examples, some are informative and some show just how confusing chemistry can be (all are a bit naughty and ignore solvent):

An example of a SN1 substitution.
The substitution of a tertiary bromide is invariably an SN1 process (not withstanding the cheeky example of an α-bromocarbonyl earlier or the possibility of addition/elimination HERE), especially if a weak nucleophile, such as water, is the reactant. Elimination of the bromide anion to give a relatively stable tertiary carbocation is proceeded by addition of water to the highly electrophilic cation. Finally, proton transfer gives the product. SN2 substitution is disfavored by the steric bulk of a tertiary halide that hinders approach of the nucleophile to the backside of the C–Br bond. The weak, neutral nucleophile, does not attack, discouraging SN2 and allowing SN1 to be favored.

An example of a SN2 substitution.
The second example is equally clear cut, but this time it favors SN2 substitution. The substrate is a primary halide and there is a strong, anionic, nucleophile. This attacks the unhindered primary position can kicks out the leaving group in a single step. Formation of a carbocation is disfavored as primary carbocations are frequently unstable.

The effect of acid on the reaction of a tertiary alcohol.
Alcohols are poor leaving groups, and do not readily undergo substitution reactions. They must be activated in some way before a reaction will occur. This is demonstrated in the example above. If you mix two alcohols together nothing will happen. Both alcohols are unreactive. There is no SN1 reaction as there is no good leaving group. There is no SN2 reaction as there still isn't a good leaving group or a good nucleophile. But, if you add a drop of acid, then an ether will start to form. What has changed? The acid can protonate the alcohols. When it protonated the tertiary alcohol an oxonium ion is created. This can be eliminated to give a stable carbocation and SN1 substitution occurs (mechanism in the first example of this section). The primary alcohol is also protonated but it will not undergo dissociation to give a primary carbocation as these are not stable.

Conversion of an alcohol into an alkyl halide.
All methods of substituting alcohols involve activation or derivatization. So far, you have seen the use of acid to make an oxonium ion and the formation of sulfonate esters. Two of the most common methods for preparing alkyl halides from alcohols involve a single reagent but still involve activation. Alkyl chlorides can be formed with thionyl chloride [S(O)Cl2] while alkyl bromides can be formed with phosphorus tribromide (PBr3). Both follow a similar mechanism, and I have shown it for bromide formation. The first step is a nucleophilic displacement with the alcohol attacking the reactant. This activates the alcohol and creates the nucleophile. In this case, a strong P–O bond is formed along with the bromide anion. SN2 substitution is favored as it is a primary alcohol and the bromide is a reasonable nucleophile. Phosphorus oxygen bonds are strong and this makes a good leaving group.

Substitution of allylic bromides (leaving group next to a alkene).
Some functional groups promote substitution reactions, making the reaction easier and faster, but it is not always clear which mechanism is in operation. A classic example involves the substitution of a leaving group α to an alkene or in the allylic position. The presence of the alkene π system can encourage either mechanism. If a poor nucleophile is used, the reaction could conceivably be SN1 as the carbocation intermediate is delocalized and this bestows a degree of stability. If the leaving group was secondary or tertiary then SN1 would be even more favored. Alternatively, a strong nucleophile would encourage SN2 substitution. The alkene can stabilize the electron deficient carbon of the transition state and this means the reaction will be faster than normal. Annoyingly, there is a third mechanism that could operate with this system. SN2' (chemists say "SN2 prime") occurs when the nucleophile attacks the alkene, which then moves to kick out the leaving group, but that is a discussion for another day!

Synthesis of a MOM-protected alcohol by an SN1 process. Acetal formation.
The reaction of methyl chloromethyl ether (MOM-Cl) initially looks as if it should be SN2 but is actually SN1. The key it identifying the mechanism is recognizing that lose of the leaving group leads to a stabilized carbocation due to delocalization of the oxygen lone pair. If you can form a stabilized carbocation the reaction is most likely to be SN1.
Conclusion
There are two common mechanisms for substitution at a saturated carbon atom. These are SN1 and SN2 substitution. These differ by the number of molecules in the rate determining step. The rate of an SN1 substitution is determined the substrate only. The reaction is first order and occurs with two discrete steps, the first is dissociation of the leaving group to give a carbocation, and the second is addition of the nucleophile. If the leaving group is on a stereocentre, the reaction often occurs with lose of stereochemical information. The other mechanism, the SN2 substitution is bimolecular with both the substrate (electrophile) and nucleophile influencing the rate of reaction. It is a single step process with all bonds being made and broken at the same time. The reaction occurs with inversion of stereochemistry (when this is a factor). At undergraduate level, the most important factor for determining whether a reaction proceeds through an SN1 or an SN2 mechanism is whether the substrate can form a stable carbocation. If the leaving group can depart to give a stable carbocation (either stabilized by hyperconjugation or delocalization), the reaction will favor SN1. If it can’t, then the reaction is more likely to be SN2. Other factors, such as solvent, can play a role but they tend to be less important.
A table is included below that tries to summarize the most common factors influencing the mechanism of substitution.
It should always be remembered that chemistry is never as simple as represented at undergraduate level. Each of the factos I have discussed has a crucial role and all must be considered. Mechanism are on a scale with the majority reactions occurring in between the two extremes outlined above. This means that the reaction may well occur in a single step (like SN2) but that the leaving group may start to depart (the bond get longer) before the nucleophile attacks (resembling SN1). There are also other substitution mechanisms and other ways of achieving the same overall transformation. This is what makes chemistry so frustrating and so much fun. Enjoy.

Summary of factors influencing mechanism of reaction. It is important to remember that these are not rules and that all factors must be considered. Additionally, it does not cover all eventualities, you could have a secondary allylic position or the dreaded neopentyl group, a primary leaving group that is a poor substrate for both SN1 and SN2.