Nucleophilic Addition to Aldehydes & Ketones
Introduction
The carbonyl group (or carbonyl bond) is shorthand for the carbon oxygen double bond or C=O. It is not a functional group in its own right but is a component of many functional groups. The majority of carbonyl containing compounds contain RC(O) or acyl fragment. This comprises the carbonyl group and R is an alkyl group (although, I am somewhat cheekily throwing in aromatic rings as well even though these are benzoyl groups if R = Ph). The other, so far, unspecified substituent on the carbonyl group determines the functional group (see summary HERE). This summary is interested in aldehydes (substituent = H) and ketones (substituent = carbon group).
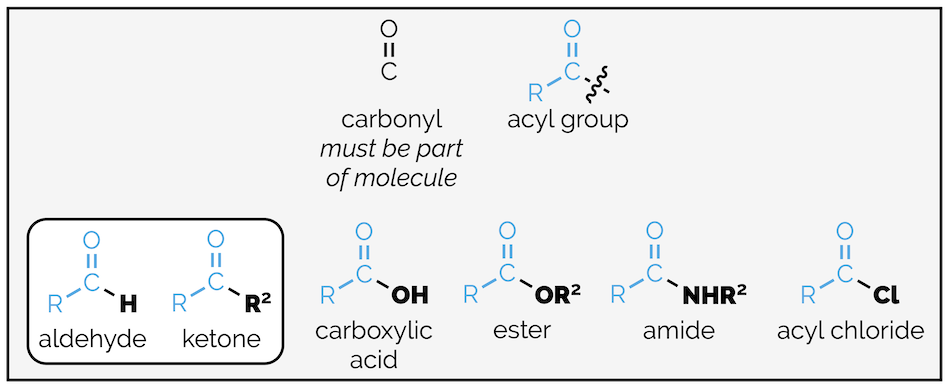
The carbonyl group, the acyl group and functional groups containing the carbonyl group.
Nucleophilic addition to the polarized carbonyl group is a key to the reaction mechanism. It is a cornerstone of organic synthesis, being used to form both C–C and C=C bonds. It is vital in biology, where formation of cyclic (hemi)acetals in carbohydrates, esterification, and ester hydrolysis in the chemistry of lipids, and, of course, amide (peptide) formation are all crucial processes.
The simplest reaction is nucleophilic addition to an aldehyde or ketone followed by protonation to give a hydroxyl-containing compound (-OH), such as hemiacetals, cyanohydrins or alcohols. In subsequent summaries, this in addition product will only be an intermediate in longer reaction mechanisms that ultimately lead to various substitution reactions but for now, I’ll stick to the top row of the diagram below:
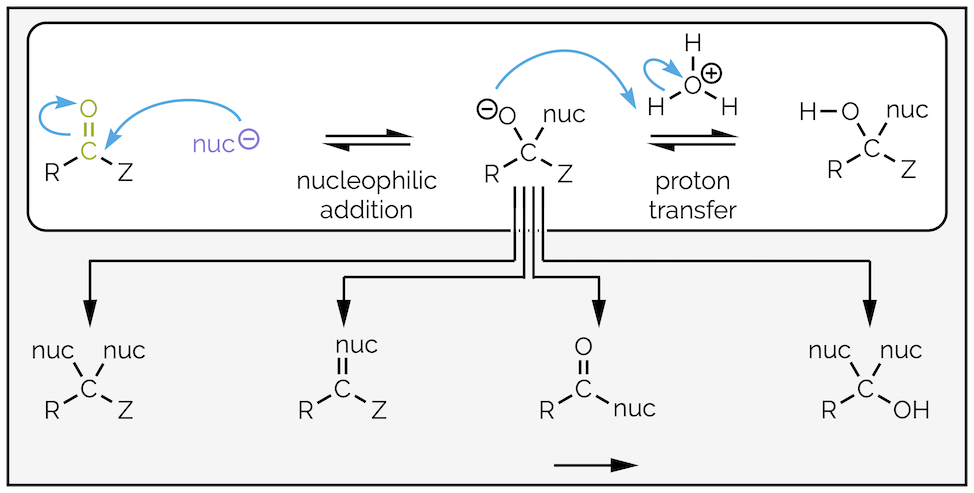
The tetrahedral intermediate formed on addition of a nucleophile to a carbonyl-containing compound can lead to a number of different functional groups depending on the nature of the nucleophile or carbonyl substituent (Z). In this summary, I am just interested in a single addition followed by protonation (white box).
Nucleophilic Addition
The carbonyl group is strongly polarized by the electronegative oxygen atom. This is further amplified by delocalization. It is possible to draw a second resonance structure with a formal positive charge on the carbon atom and the corresponding negative charge on the oxygen atom.
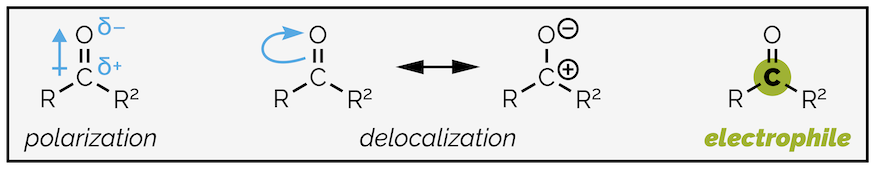
Polarization and delocalization of the carbonyl group make the carbon highly electrophilic.
Polarization of the carbonyl group has two effects. It makes the C=O double bond stronger and shorter than an alkene C=C. The electrostatic attraction between the partially charged carbon and oxygen can be thought as adding a degree of ionic bonding to the group. While the bond is stronger, it is also more reactive with the electrophilic carbon readily attacked by nucleophiles.

Comparison of the physical properties of a carbonyl C=O bond and an alkene C=C bond.
The strength of the carbonyl group means that it is often called a thermodynamic sink. Its formation is energetically favorable, and it is the driving force pushing many reactions forwards (as you will see in subsequent summaries). The strength of this bond is one of the many reasons nucleophilic addition to aldehydes and ketones can be reversible.
Taking things further: The carbonyl group and infrared spectroscopy CLICK HERE.
This summary focuses on nucleophilic addition to aldehydes and ketones. Reactions that involve substitution of the oxygen (condensation reactions), or those involving functional groups with an addition heteroatom (acyl substitution reactions), will be the topic of subsequent summaries (it wouldn’t be a summary if I try to cover it all).
The general reaction for nucleophilic addition to aldehydes and ketones is:

The general reaction for nucleophilic addition (of a nucleophile called nuc-) to an aldehyde or ketone followed by protonation. The groups acting as a nucleophile are in purple while the electrophiles are in green.
Nucleophilic addition to an aldehyde or ketone is a two step process. The first is nucleophilic addition to the carbonyl group. To understand this reaction, let’s look at it in detail. A logical approach is to identify which bonds have been made and those that are broken. Then identify the nucleophile and electrophile before adding the curly arrows that show how the electrons are redistributed.
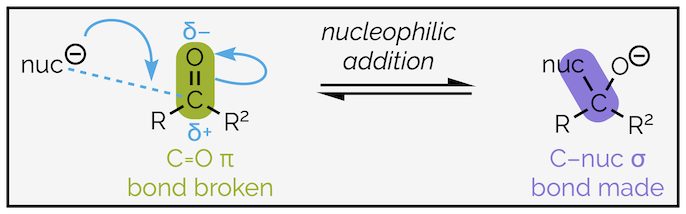
The first step in nucleophilic addition. The nucleophile attacks the electrophilic carbonyl group to form a new bond and the tetrahedral intermediate.
The nucleophile will be an electron rich species that can either be negatively charged (as drawn) or neutral. It will have either a lone pair of electrons or a high energy, polarized bond, with the partial negative charge on the nucleophilic atom. The electrons are attracted to the partial positive charge of the carbonyl and a bond is created between the nucleophile and the carbon atom. This can be shown by a curly arrow, the tail of which will starts with either the lone pair of electrons or the bond of the nucleophile. The head ends on the new bond. It should be halfway between the nucleophile and carbon but is normally depicted closer to the carbon atom. The neutral carbon atom of the C=O cannot have ten valence electrons (five bonds), so two electrons must be taken away (or leave). This is achieved by breaking the carbonyl π bond and moving the electrons to the electronegative oxygen atom. Again, this can be shown by a curly arrow, the tail of which starts where the electrons are in the original diagram (the bond) and its head ends where they will be in the second diagram (the oxygen atom). This is an example of nucleophilic addition mechanism, coupled with bond breaking (see summary HERE).
This addition results in the trigonal planar or sp2 hybridized carbon of the carbonyl group becoming tetrahedral or sp3 hybridized. As shorthand, the intermediate is known as the tetrahedral intermediate. This intermediate will be key to all the reactions of the carbonyl group that I'll be covering over the next couple of summaries.
The second step is proton transfer:
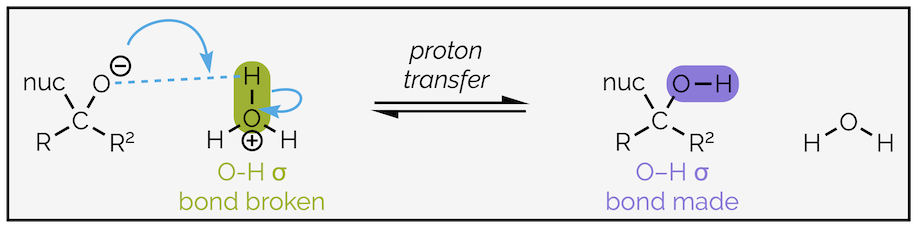
Proton transfer from a hydronium ion to the tetrahedral intermediate (anion).
Identifying which bond is made (O–H) and which is broken (O–H) helps you to draw the mechanism. Any bond that breaks will have a curly arrow starting in the middle, while any bond being formed will have the head of curly arrow indicating where it should be drawn. Identifying the nucleophile in this example is simple. It is the negatively charged species while the electrophile is the positively charged species. It is then a case of drawing an arrow from the nucleophile to create the new bond (a nucleophile always attacks an electrophile to form a new bond), then a second arrow to break the old H–O bond.
Proton transfer can be an internal (intramolecular) process if a neutral nucleophile with an X–H bond was used, or it can occur when an acid or proton source is added at the end of the reaction as shown above. The addition of a proton source is often called quenching the reaction or performing a(n acidic) work-up. There will be examples of both internal and external proton sources in the following section.
The Reversibility of Nucleophilic Addition
The carbonyl group is a reactive electrophile, and is readily attacked by a nucleophile, but it always try to reform if possible. Why?
Nucleophilic addition is a favorable process as a strong σ bond is formed at the expense of a weak π bond. This drives the reaction forwards. The reverse process, elimination of the nucleophile to reform the carbonyl group can also be a favorable process and this can lead to an equilibrium being established. The stability of the carbonyl bond drives the reverse reaction but it is only possible if there is a leaving group on the tetrahedral intermediate. A leaving group is an atom or group of atoms capable of taking away two electrons in the form of either a stabilized negative charge or lone pair of electrons. The leaving group can either be the original nucleophile as shown below or it can be one of the substituents on the carbonyl group (R or R2). The latter will be dealt with in a separate summary.

Nucleophilic addition to form the tetrahedral intermediate (blue arrows on the left) or elimination, kicking out the nucleophile as a leaving group (the red arrows on the right).
The curly arrows for the elimination of the opposite as those for addition. A lone pair of electrons on the oxygen is shared between the oxygen and carbon atom to create the π double bond. This is shown by an arrow whose tail starts on the lone pair of electrons (often simplified to starting on the negative charge that indicates that there must be an extra lone pair) and moves to the center of the C–O bond to indicate that a second bond is being created. The carbon cannot have ten valence electrons or five bonds so two are taken away by the leaving group (the nuc). The arrow showing this starts in the middle of the bond that will be broken and goes to the atom that will gain a lone pair of electrons.
The position of the equilibrium can be predicted by inspecting the two basic species, the nucleophile (nuc– or LG– (abbreviation for Leaving Group)), and the tetrahedral intermediate (O–). Reactions favor formation of the most stable, least reactive or weakest base. This means if nuc– is a strong base, then addition is favored. Otherwise, if nuc– is a weak base, elimination and formation of the carbonyl is favored. Which side is favored can be determined by comparing the pKa of the conjugate acid or pKaH. The side of the reaction that has the species with the lowest value of KaH, the stronger conjugat acid or weakest base, will be favored (see the summary HERE if you don't understand this concept).
Taking things further: The Orbital Approach to addition to the Carbonyl Group CLICK HERE.
The Reactivity of Aldehydes and Ketones
Aldehydes are more reactive than ketones for two reasons. Aldehydes are more electrophilic. An alkyl group is electron donating (think about the stability of cations and hyperconjugation). An aldehyde has one alkyl group pushing electrons onto the partially positive carbon atom and reducing the electrophilicity. A ketone is less electrophilic as two alkyl groups reduce the partial positive charge.

A carbonyl group is highly polarized and electrophilic on the carbon atom. Adding electron donating groups pushes electron density onto the carbon and reduces the electrophilicity. Two groups have a bigger effect than a single group.
The second argument involves sterics. The nucleophile approaches the carbonyl group along an angle of 107° (the Bürgi-Dunitz angle). This means it has to pass over the molecule. An aldehyde has a hydrogen substituent that does not influence the approach (in fact, the hydrogen allows the nucleophile to minimize interaction with the rest of the molecule). A ketone has two carbon-based substituents. These interact with the approaching nucleophile and will slow the rate of reaction. Ketones are less reactive.
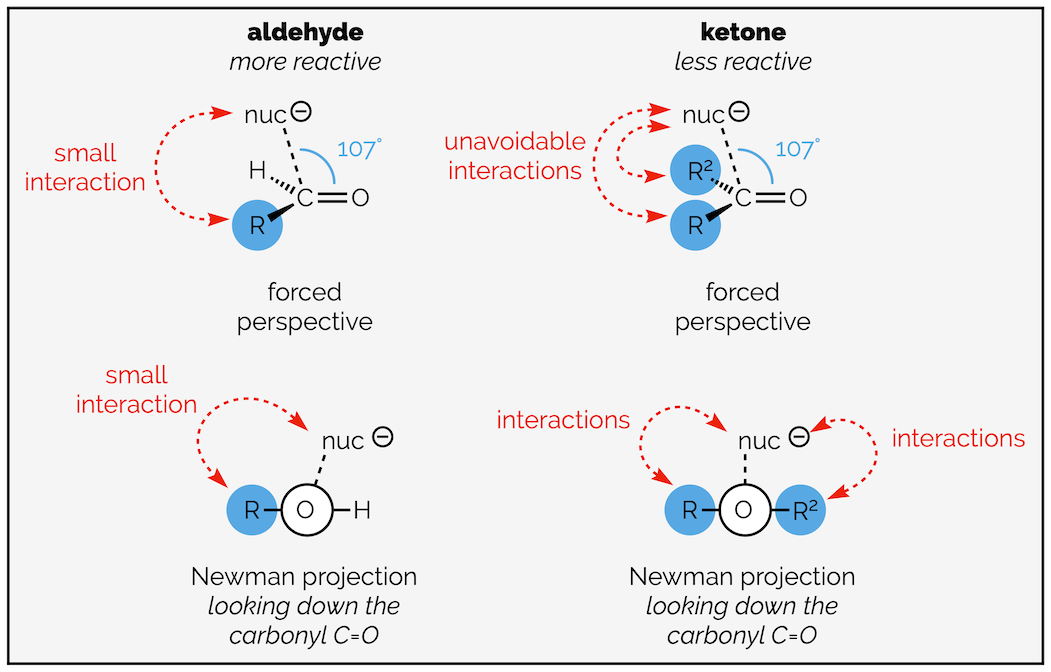
The steric argument for the reduced reactivity of ketones; some form of interaction between incoming nucleophile and the bulk of the molecule is unavoidable.
Irreversible Addition to Aldehydes and Ketones
The nucleophilic addition of organometallic reagents, such as Grignard (organomagnesium) reagents R–MgX or organolithium reagents R–Li, to an aldehyde or ketone is irreversible as shown for the addition of butyllithium to acetone.
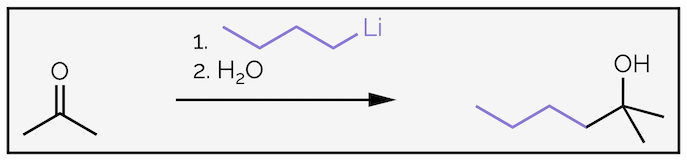
The addition of butyllithium to acetone followed by the addition of water gives an alcohol.
The carbon atom is more electronegative than the metal causing a highly polarized bond with the electron density closer to the carbon atom. The carbon atom is the nucleophile. Many organic textbooks simplify these reagents to anions. The two versions are below, and are equally correct for most introductory chemistry courses:
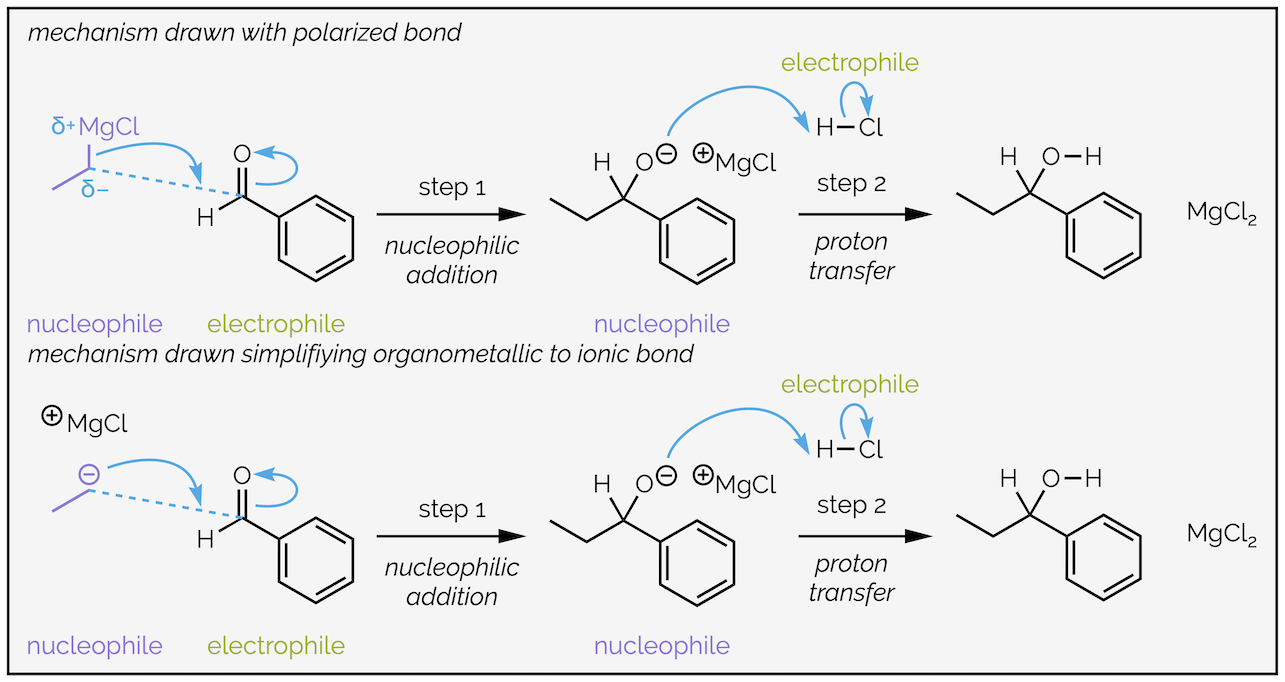
Two versions of the mechanism for the addition of a Grignard reagent (organomagnesium nucleophile). Both are simplifications of what is “really” happening as the reactions almost certainly involve at least two equivalents of the Grignard reagent and solvent molecules.
The example above involves a Grignard reagent or organomagnesium reagent. For most introductory organic chemistry courses organomagnesium and organolithium reagents are interchangable. The reaction involves the formation of a C–C bond between the nucleophilic organometallic reagent and the electrophilic carbonyl group. The tail of the arrow will start from the nucleophile, either the two electrons of the polarized bond or the two electrons of a lone pair on carbon. The head of the curly arrow must end where the new bond will be formed.
Addition to the carbonyl group requires the C=O double bond to break and take two electrons away so that the carbon maintains eight valence electrons. The electrons flow towards the electronegative oxygen atom, which gains a lone pair of electrons (and also maintains eight valence electrons). These two arrows make up step 1, the nucleophilic addition.
Step 2 is the addition of a proton source (proton transfer), in this case hydrochloric acid. The alkoxide or oxygen anion is the electron rich nucleophile and attacks the proton with electrons flowing towards the electronegative chlorine.
The proton source must be added in a separate step both in terms of the reaction mechanism and in a laboratory setting (this is known as “quenching the reaction” or is part of the “work-up”. This is shown on a reaction by either writing 1. & 2. before each reagent or “then H+ (or whatever the proton source is)”.

Two, of the many, different ways of writing the reaction of an aldehyde or ketone with an organometallic reagent. The key is that the organometallic reagent and the proton source cannot be mixed together.
The steps are separate as organolithium and Grignard (organomagnesium) reagents will react with good proton sources such as acid or water. This destroys them. As a generalization, any nucleophile that is sufficiently basic to add irreversibly (see below) will be sufficiently basic to react with water.
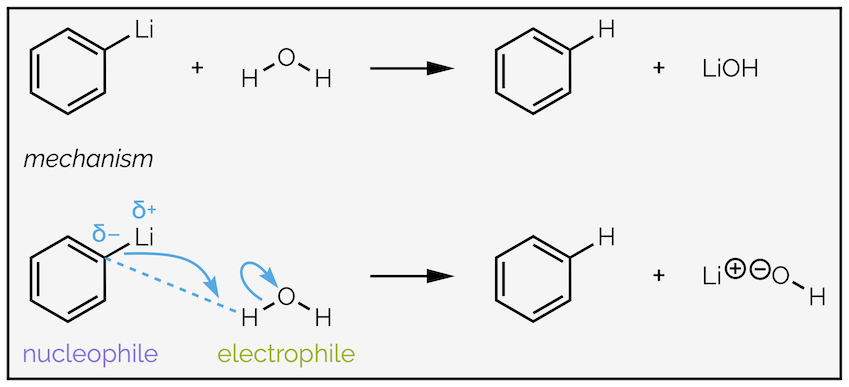
Proton sources can destroy many organometallic reagents.
The addition is irreversible as there is no leaving group on the tetrahedral intermediate. If you consider the addition of methyllithium to benzaldehyde as shown below. Addition results in the formation of an alkoxide. For the carbonyl group to reform, a lone pair of electrons on the oxygen must be shared with the carbon. To avoid ten valence electrons (impossible in the second row of the periodic table), two electrons must be taken away. The possible leaving groups are Ph–, H– or CH3–. None of these are more stable or favorable than the alkoxide. If you compare the pKaH of the alkoxide against each of these you will see it is by far the most acidic species or the weakest base. The reaction always favors the side with the weakest base (or creates the weakest acid).

The addition of an organometallic reagent to an aldehyde is irreversible as there is no suitable leaving group to allow the carbonyl bond to reform. This can be seen from comparison of the strength of the bases (or conjugate acids).
The pKa of the conjugate acid of the methyl anion is over three times that of the alkoxide (48 vs 15.4). This means it is a weaker acid or a stronger base. Its formation is not favored. It is the same situation for both the hydride and the benzene anion. All of them of far less stable than the alkoxide so will not form. The reaction is irreversible. The only solution is to add a proton source and form an alcohol.
Aldehydes and ketones can be reduced to alcohols by an irreversible nucleophilic addition to the carbonyl bond. The two most common reagents are lithium aluminium hydride and sodium borohydride. These behave in the same manner as the organometallic reagents.

The reduction of aldehydes and ketones with lithium aluminium hydride or sodium borohydride. Both reagents will reduce both aldehydes and ketones.
The reaction mechanism for the two reagents is different when you really dig into it but for most undergraduate courses they can be considered the same, and are simply examples of nucleophilic addition to the carbonyl group. Below, I have shown the mechanism for the lithium aluminium hydride reduction of acetophenone.

The mechanism for the lithium aluminium hydride reduction of a ketone.
There is no difference between this reaction and those of the organometallic reagents above. The negative charge indicates that the aluminium hydride anion is electron rich and is the nucleophile. But don’t let the charge confuse you. There is no lone pair of electrons on the aluminium so a bond must be the nucleophile, and while the formal charge gives the aluminum the negative charge it is the polarization of the bond that is key to determining which atom adds. Hydrogen is more electronegative than aluminium so it has the higher electron density and it behaves as the nucleophilic atom. This mirrors the polarization in an organometallic reagent.
The curly arrow starts with the the two electrons of the Al–H bond. Its head points to the new bond formed between the hydrogen and carbon. As with all nucleophilic additions to the carbonyl group, formation of the new bond is accompanied by breaking the C=O double bond. This gives the tetrahedral intermediate. In a separate step, a proton source is added to allow formation of the desired alcohol.
Lithium aluminium hydride reacts violently with water. The proton transfer must be performed after reduction and will always be depicted as the second set of reagents (or with the text ‘then proton source’).

The first step in the reaction of lithium aluminium hydride and water. More reactions occur after this, none desirable to most organic chemists.
The reaction is irreversible. Once the tetrahedral intermediate has been formed the carbonyl group cannot be regenerated as there is no suitable leaving group. In the example above, reforming the carbonyl group would require either a hydride H- to leave, a methyl anion CH3– to leave or the benzene anion C6H5– to leave. All are stronger bases than the alkoxide and their formation is disfavored. The pKaH are listed in the previous organometallic example.
Many textbooks simplify the addition of hydride reducing reagents to the equation below. This is the same as simplifying an organometallic reagent to a carbanion and metal cation. The advantage of such a simplification is that it is easy to spot the nucleophile and it it makes the similarities between all nucleophilic additions to a carbonyl group very apparent. The disadvantage is that it is not true!

The common simplification for hydride reducing regents. It is not correct!
Hydride, H–, is not a nucleophile. Sodium hydride, NaH, is an ionic compound that is the closest we have to H–. It is a strong base. Hydride, H–, is a strong base because the charge is not stable (if this isn't ringing any bells, read the summary on predicting acids and bases HERE). Hydrogen isn't less electronegative than carbon and it is small. It cannot stabilize the charge. Hydride is not a good nucleophile due to its size. The more polarizable a reagent, the more nucleophilic it is. Polarizability occurs when charge can be unevenly distributed in a molecule. It is greatest in big molecules. Hydrogen is the smallest atom and the charge is concentrated in a small area so is not polarizable.
More complicated explanation:
Alternatively, you can consider the orbitals involved in a reaction. Reactions occur when there is optimum orbital overlap. The maximum overlap occurs when the orbitals are of similar sizes. A hydride has a full 1s orbital. This is the smallest orbital. It will overlap effectively with the C–H σ* antibonding orbital as this was made from a hydrogen 1s orbital. Hydride is a good base. There is poor overlap between a hydride 1s orbital and the large diffuse 2p character of a C=O π* antibonding orbital. Thus the hydride doesn't add to a carbonyl group.
Sodium borohydride NaBH4, is not as reactive as lithium aluminium hydride. It is a poorer nucleophile. Again this can be explained if you consider that nucleophilicity is related to polarizability. Boron is smaller than aluminium and BH4- is less polarizable than AlH4- so is less nucleophilic. The B–H bond is less polarized than an Al–H bond. The difference in electronegativity between boron and hydrogen is 2.20 - 2.04 = 0.16 while the difference between aluminium and hydrogen is 2.20 - 1.61 = 0.59.
As sodium borohydride is less reactive than lithium aluminium hydride, a protic reaction solvent can be employed in reductions. The solvent then acts as the proton source required for the proton transfer step. In fact, these reactions are commonly performed in methanol or ethanol.
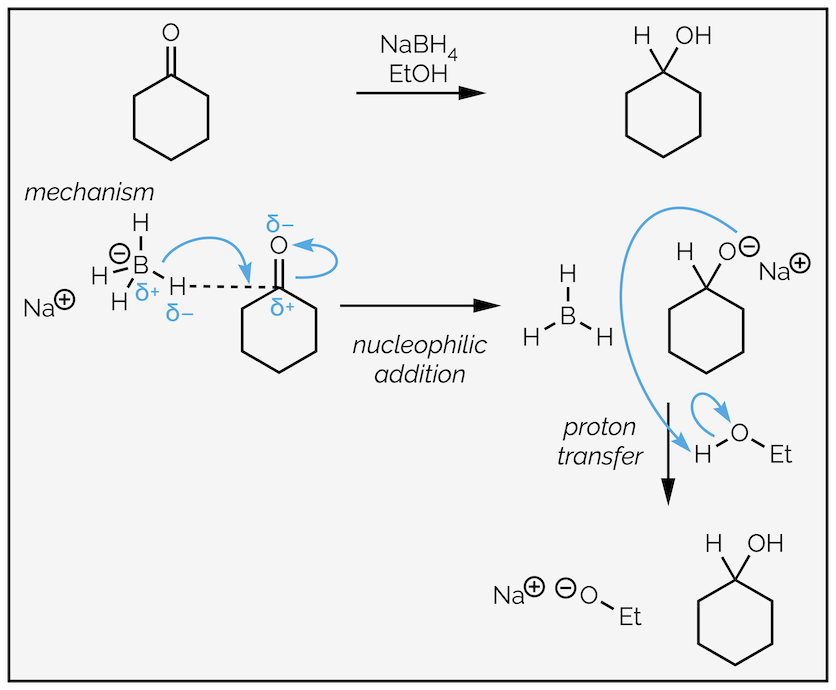
Reduction of a ketone with sodium borohydride. The reduced nucleophilicity of sodium borohydride permits the use of protic solvents that also act as the source of protons in the second step of the nucleophilic addition mechanism.
Other than the change in solvent, the reaction is identical to before and is an example of nucleophilic addition followed by protonation. The hydrogen atom is the nucleophile due to the polarity of the B–H bond. The two electrons of the B–H bond are donated to the electrophilic carbonyl group. This is shown by a curly arrow starting with the bond and ending where the new C–H bond will be. The carbonyl π bond is broken. The curly arrow shows the movement of the bond to become a lone pair of electrons on the electronegative oxygen. The second step is the proton transfer. The alkoxide attacks the proton of the solvent to give an alcohol and a new alkoxide. The more astute might note that this should be an equilibrium … it is actually a simplification. The new alkoxide or the solvent will react with the borane produced in the first step but that is a story for a different day.
Taking things further: The Orbital Approach irreversible nucleophilic attack on the carbonyl Group CLICK HERE.
Reversible Addition to Aldehydes and Ketones
The addition of water to aldehyde or ketone can occur to give a functional group known as a hydrate as shown below:

Water can add to an aldehyde by nucleophilic addition.
Funnily enough, the mechanism is the same as any other nucleophilic addition to the carbonyl group. Water is the nucleophile, a lone pair of electrons is donated to create a new bond between the oxygen and the carbon atom. With nucleophilic addition to the carbonyl there must be breaking of the π bond to take two electrons away. This gives the tetrahedral intermediate as shown:

The mechanism for hydrate formation is identical to that of the addition of organometallic reagents with one important exception, the reaction is now reversible.
After the addition, the second step is proton transfer. There are a number of ways of drawing this process. All are allowable. Above, I have drawn a two-step intermolecular (occurs between two separate molecules) proton transfer. First a molecule of water deprotonates the zwitterionic (a neutral molecule that has both a formal negative and formal positive charge) tetrahedral intermediate. The second step, uses the hydronium species to quench the negative charge. The two other proton transfer mechanisms are the intramolecular variant, in which the proton simply skips from one atom to another, and a concerted intermolecular transfer.

Alternative mechanisms for proton transfer (all are correct so don't panic).
The nucleophilic addition of water to an aldehyde or ketone is slower than the addition of an organometallic reagent or metal hydride reducing reagent. Water is less nucleophilic. It is less polarized, with the electronegative oxygen atom holding onto the lone pair of electrons more tightly. It is a poorer electron donor. The structure of the carbonyl also influences the rate of reaction, with more sterically demanding groups further slowing the addition. In fact, only aldehydes normally give any appreciable quantity of hydrate.
The key difference to the previous additions is the reaction is now reversible. There is now a leaving group on the tetrahedral intermediate, and the carbonyl group can reform through an elimination reaction:

Elimination from water from the tetrahedral intermediate.
A curly arrow starting from the lone pair of electrons on the oxygen represents the formation of the π bond. Two electrons must be removed from the carbon and the C–O σ bond is broken to give water. Comparing the pKa of the conjugate acids (pKaH) shows that elimination is the favored reaction. The pKa of an alcohol (left-hand side) is approximately 15.5 while that of the hydronium ion (the conjugate acid of water on the right-hand side) is 0. Water is the weaker base and its formation is favored.
Alcohols can participate in nucleophilic addition to aldehydes and ketones in exactly the same manner. Water and alcohols are both neutral oxygen-centred nucleophiles so you should expect them to behave the same. Both use a lone pair of electrons to attack the carbonyl group. Both lead to an equilibrium that normally favors the carbonyl group rather than the hydrate, or, if an alcohol adds, the hemiacetal.

Formation of hemiacetals by the nucleophilic attack of an alcohol on an aldehyde or ketone. Mechanism shown below the overall reaction.
Just like the formation of hydrates, the equilibrium normally favors the aldehyde or ketone but there is one important exception and that arises with the formation of cyclic hemiacetals. This occurs when the alcohol and carbonyl group are in the same molecule.
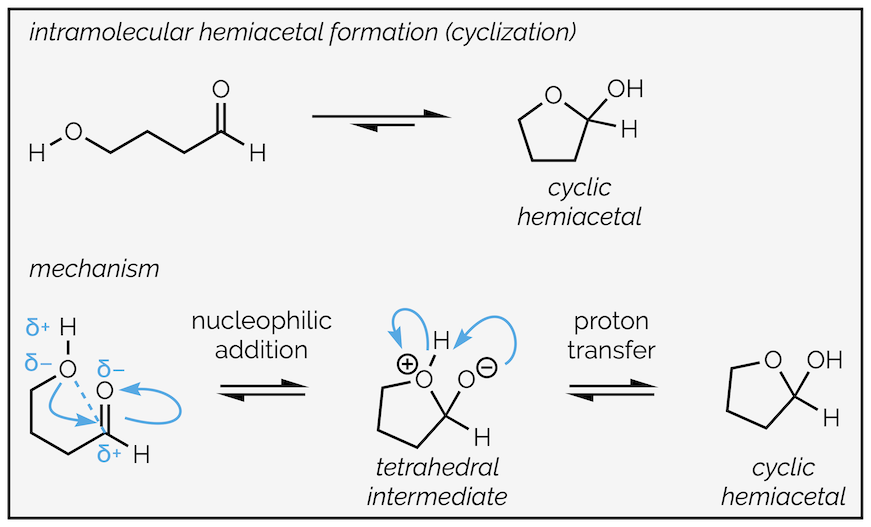
Intramolecular hemiacetal formation. Occurs when alcohol and carbonyl are in the same molecule. The resulting cyclic hemiacetal is more stable than the acyclic version.
The importance of this is found in biology and the structure of sugars such as glucose:
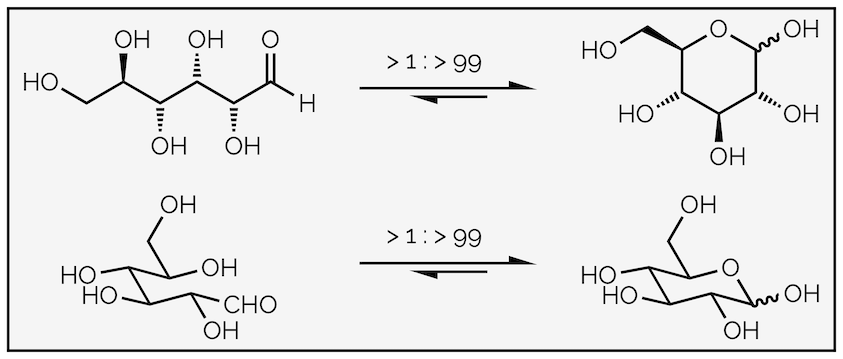
Intramolecular hemiacetal formation is favored for many carbohydrates or sugars. This diagram shows two different representations of glucose cyclizing to form the hemiacetal. The wiggly line at the hemiacetal (anomeric) position indicates that I'm not defining the stereochemistry. This is because that would be a summary in its own right.
Hemiacetals can also be formed under catalytic conditions. Reacting a mixture of aldehyde or ketone with an alcohol and a sub-stoichiometric quantity of alkoxide gives the same equilibrium as above. The alkoxide speeds up the reaction and is not consumed in the reaction (or, more accurately, is regenerated during the reaction). The alkoxide is more nucleophilic than the alcohol. The negative charge indicates it is more electron rich. It participates in nucleophilic attack to form a tetrahedral intermediate. Proton transfer occurs between anion of the hemiacetal and the alcohol to regenerate the nucleophilic alkoxide.
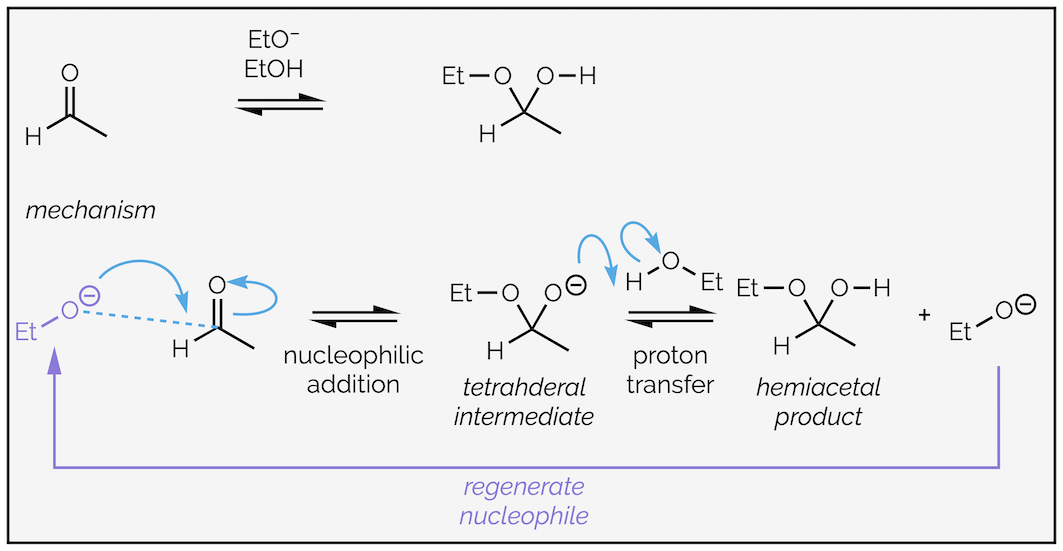
The base-catalyzed formation of hemiacetals. This mechanism works for the formation of hydrates as well (as they are the same reaction for all intents-and-purposes).
The equilibrium is now between to oxygen anions. Comparing the pKaH of the left-hand side (ethanol pKa = 15.5) and the right-hand side (an alcohol-like species pKa around 15.5) there is barely a difference. Neither side is particularly favored.
The last nucleophile I want to look at in this summary is the cyanide anion. This permits the reversible formation of a C–C bond. The mechanism is exactly the same as all the other examples in this summary. It starts with nucleophilic addition to the carbonyl group, which leads to the tetrahedral intermediate. This is followed by proton transfer to give a compound known as a cyanohydrin. The cyanide anion is a weak base, the pKa of hydrogen cyanide or cyanide gas is 9.2, making it slightly more acidic than phenol. This means reactions can be conducted in the presence of an acid although you should be careful not to synthesize cyanide gas.

Cyanohydrin formation and mechanism.
The position of the equilibrium generally favors the carbonyl starting material (the cyanide anion is a weaker base than an alkoxide (pKaH 9.2 versus 15.5)). The structure of the carbonyl compound has the greatest influence on the position of the equilibrium with aldehydes giving more cyanohydrin than ketones. This can be explained by thinking of steric hindrance. Cyanohydrins have a tetrahedral carbon compared to the trigonal planar carbon of the starting material. This means the bond angles have decreased from 120° to 109° and the substituents on the carbon are closer. This is less important in aldehydes as one of the substituents is a small hydrogen atom.
Taking things further: The Orbital Approach to the addition of water or cyanide to an aldehyde CLICK HERE.
Catalysis
There are two possibilities for catalyzing the addition of a nucleophile to an aldehyde or ketone. You can either increase the strength of the nucleophile or the electrophile. The use of a base to promote the addition of either water or an alcohol to give the hydrate or hemiacetal is an example of enhancing the nucleophilicity of the reaction or base catalysis. The opposite process is enhancing the electrophilicity of the carbonyl group. This can be achieved by acid catalysis.
Base catalysis was covered in the section hemiacetal formation above.
Adding acid to certain reactions results in protonation of carbonyl group. This enhances the polarization of the carbonyl bond and makes the carbon atom more electrophilic. This, in turn, enhances the rate of nucleophilic addition.

Nucleophilic attack on the polarized carbonyl group versus protonation to enhance the electrophilicity, followed by nucleophilic addition.
Acid catalysis adds an addition protonation step to our general mechanism. In the first step, the carbonyl group acts as a nucleophile. Every polarized bond has an electrophilic end (partially positive), and a nucleophilic end (partially negative). As long as the partially negative end can donate electrons it can be a nucleophile. The carbonyl oxygen has two lone pairs of electrons and one of these can attack an acid to form an oxonium ion. The positive charge causes greater polarization of the carbonyl group. It makes nucleophilic attack easier.
Acid catalyzed hydrate formation is shown below:
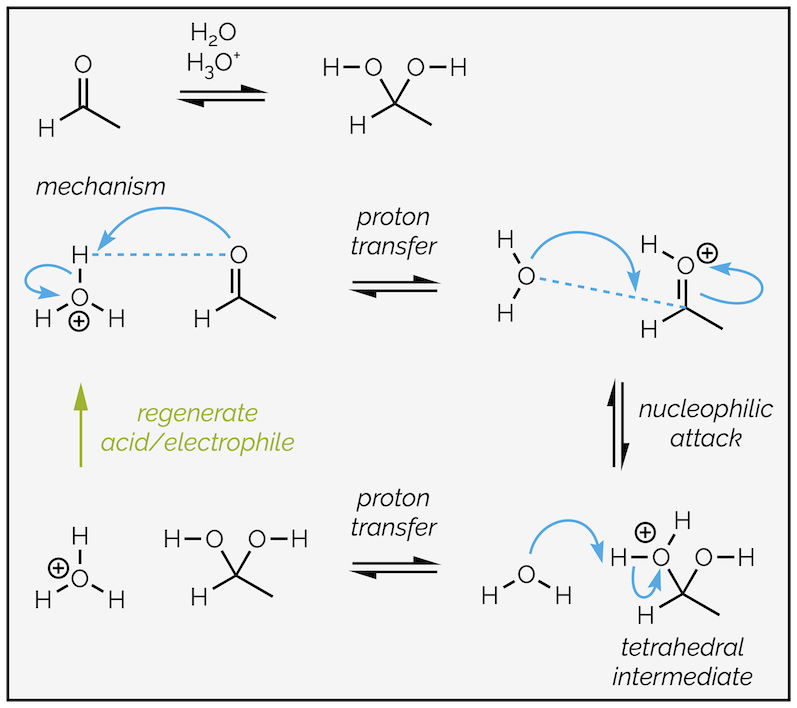
Acid-catalyzed hydrate formation. An addition step activates the aldehyde to nucleophilic attack.
There are limitations to (protic or Brønsted) acid catalysis. Firstly, the nucleophile cannot be a strong base, otherwise the predominant reaction is protonation of the nucleophile not activation of the electrophile. This leads to destruction of the nucleophilic reagent. Protic acids destroy organometallic reagents or metal hydride reducing reagents instead of activating the aldehyde or ketone. Secondly, protonation can activate the tetrahedral intermediate as well as the carbonyl group and this leads to a different reaction. The acid can protonate the hydroxyl OH group and make a good leaving group. This changes the reaction and permits the overall substitution of the oxygen atom instead of just addition. This occurs when alcohols or amines are the nucleophile and is the topic for a future summary (condensation reactions).
Conclusion
Aldehydes and ketones contain a polarized carbonyl bond. This makes them good electrophiles. Nucleophilic addition to aldehydes and ketones proceeds by a two-step mechanism that delivers a tetrahedral product. The first step is nucleophilic addition to the carbonyl bond. This is accompanied by breaking the carbonyl π bond to give the negatively charged tetrahedral intermediate. The second step is a proton transfer that quenches the negative charge to give a neutral species. This second step can occur with reagents found in the reaction mixture but often requires the addition of a proton source in a step known as the 'work-up' or 'quenching the reaction'.
Some nucleophiles, such as organometallic reagents or reducing reagents, add irreversibly to the carbonyl group. The reaction is irreversible as there is no suitable leaving group (weak base) on the tetrahedral intermediate. These reagents invariably require the addition of an external proton source to quench the reaction. Other nucleophiles add reversibly and an equilibrium between aldehyde/ketone and the product is formed. This occurs when the nucleophile is also a good leaving group (it is a weak base).
The molecular orbital representation of these reactions has been introduced as an aside for those that are interested but it is not necessary for many introductory organic courses.
Acid and base (Brønsted definition) catalysis can be employed in some nucleophilic additions. Acid catalysis cannot be used for all reactions as the acid will either destroy the nucleophile or can cause a substitution reaction to occur. Such reactions will be the subject of a future summary.